Amyotrophic Lateral Sclerosis (ALS)
Understanding ALS: Causes, Symptoms, and Latest Research Updates
Amyotrophic Lateral Sclerosis (ALS), a name that evokes the legendary baseball player Lou Gehrig, is a devastating and progressive neurodegenerative disease that relentlessly attacks the nervous system. It affects the motor neurons—the crucial nerve cells that govern voluntary muscle movement. This debilitating condition gradually robs individuals of their ability to walk, speak, swallow, and eventually, breathe. As a complex enigma that has perplexed scientists for decades, understanding ALS—its causes, its insidious symptoms, and the cutting-edge research striving for breakthroughs—is paramount. This article aims to provide a comprehensive yet accessible overview, shedding light on the scientific landscape, the challenges patients face, and the growing hope fueled by advances in research and treatment.
What is Amyotrophic Lateral Sclerosis (ALS)?
Amyotrophic Lateral Sclerosis (ALS) is a specific type of motor neuron disease. It selectively targets nerve cells in the brain and spinal cord, which are essential for controlling all voluntary muscles. The very name “Amyotrophic Lateral Sclerosis” provides insight into its pathology: “Amyotrophic” originates from Greek, meaning “without muscle nourishment,” reflecting the muscle atrophy that occurs as the nerves controlling them degenerate. “Lateral” refers to the sections in the spinal cord where these critical nerve cells are found. “Sclerosis” signifies the hardening or scarring that develops in these affected nerve pathways as they are damaged.
A Brief History: Lou Gehrig’s Disease and Charcot’s Disease
The disease gained significant public attention in the United States through the tragic diagnosis of the beloved baseball icon Lou Gehrig in 1939, which led to its common association with his name. However, ALS was first scientifically described by French neurologists Jean-Martin Charcot and his colleagues during the 1860s. In recognition of their foundational work, it is frequently referred to as Charcot’s disease in many parts of the world. These historical connections highlight the long-standing medical interest and the persistent struggle to comprehend and combat this intricate neurological disorder.
The Devastating Impact: A Neurodegenerative Condition Affecting Motor Neurons
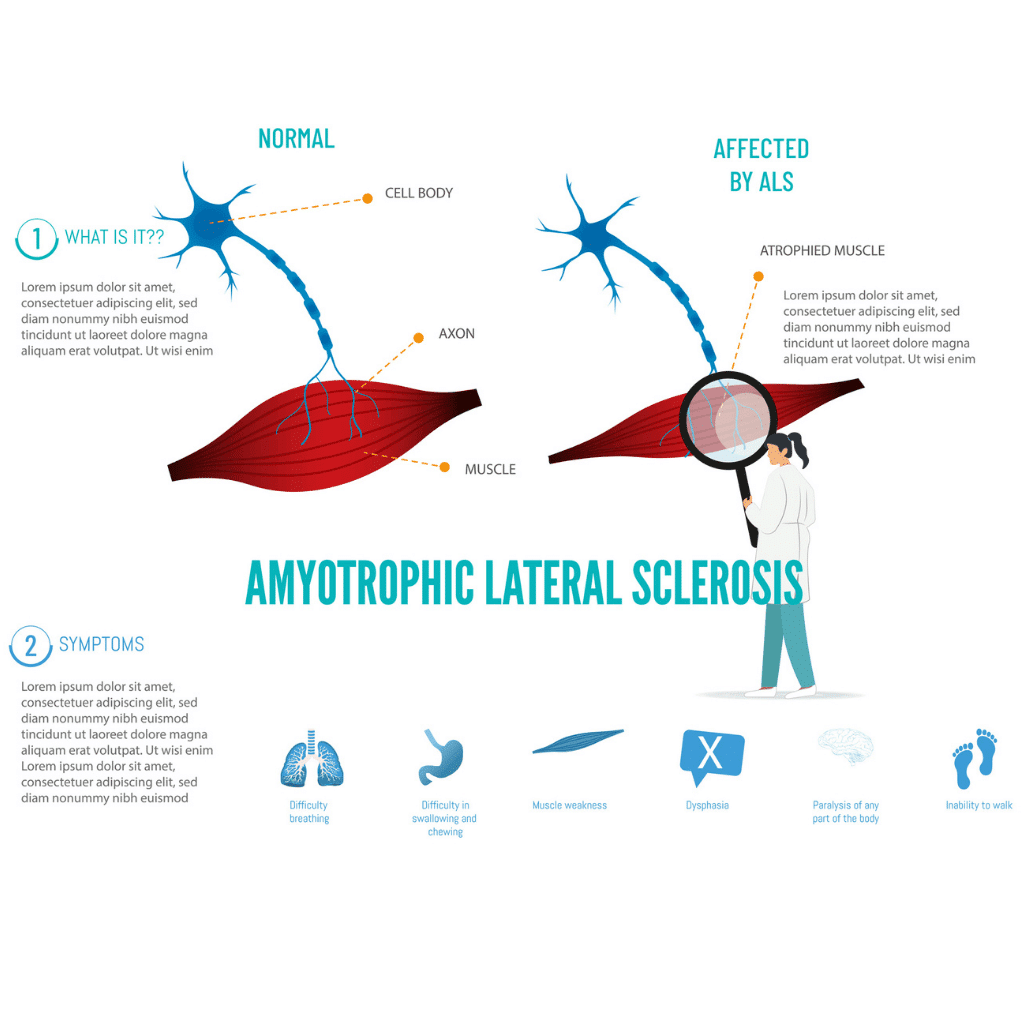
At its core, ALS is a devastating neurodegenerative condition, meaning it is characterized by the progressive death of nerve cells. In ALS, the primary cells affected are the motor neurons: both the upper motor neurons originating in the brain and controlling voluntary movement, and the lower motor neurons that extend from the spinal cord to the muscles themselves. As these motor neurons degenerate and die, the vital signals they transmit to the muscles are disrupted. This interruption leads to a cascade of effects, including progressive muscle weakness, atrophy (wasting), and ultimately, paralysis. This relentless degeneration impacts virtually all voluntary muscle functions, profoundly affecting an individual’s quality of life.
What is ALS? A Closer Look at Motor Neuron Disease
Understanding ALS inherently means understanding the critical role of motor neurons and how their degeneration defines the disease’s progression and its devastating consequences. ALS is the most common form of motor neuron disease, a group of neurological disorders that affect the nerves that control voluntary muscles.
Defining ALS: The Progressive Degeneration of Motor Neurons
Amyotrophic Lateral Sclerosis (ALS) is medically defined by the gradual and irreversible loss of motor neurons. These neurons act as the vital messengers, transmitting signals from the central nervous system—the brain and the spinal cord—to the body’s muscles, thereby initiating and controlling every voluntary movement we make. As ALS progresses, these communication pathways become increasingly damaged, leading to a breakdown in the crucial connection between the brain and the muscles. This degeneration is not confined to a single area but affects motor neurons throughout both the brain and the spinal cord, leading to widespread functional loss.
The Role of Motor Neurons: Controlling Muscle Movement
Motor neurons are absolutely fundamental to our ability to interact with and navigate the world around us. They enable everything from the delicate, fine motor skills required for tasks like writing or intricate manipulation, to the gross motor movements necessary for walking, running, and maintaining posture. They also play a role in essential involuntary actions like breathing and swallowing. When motor neurons are healthy, they efficiently transmit electrical and chemical signals that cause muscle fibers to contract, resulting in orchestrated movement. In ALS, however, the dysfunction and eventual death of these neurons cripple this essential communication system, leading to loss of function.
Progressive Nature of the Disease: From Initial Weakness to Widespread Impact
A defining characteristic of ALS is its inexorable progressive nature. Symptoms typically commence subtly, often initially affecting specific, localized muscle groups. This early muscle weakness might manifest as a slight difficulty lifting the front of the foot (foot drop), a noticeable clumsiness in the hands, or subtle changes in speech clarity. As the disease advances, the degeneration spreads to encompass more motor neurons and consequently, larger areas of the body. This relentless progression leads to increasingly severe muscle weakness, pervasive paralysis, and significant challenges with essential life functions. For individuals living with ALS, the disease’s relentless march means that over time, they will eventually require substantial assistance with nearly all daily activities, from eating and dressing to breathing.
The Enigma of ALS: Exploring Causes and Risk Factors
Despite decades of intensive research, the precise cause of Amyotrophic Lateral Sclerosis (ALS) remains unknown for the vast majority of cases, presenting a profound challenge in prevention and treatment. The disease’s complexity arises from a combination of genetic predispositions and potential environmental influences.
Sporadic vs. Familial ALS: Understanding the Two Main Categories
ALS is broadly categorized into two primary forms: sporadic and familial. Sporadic ALS is the far more common type, accounting for approximately 85-90% of all diagnoses. In sporadic ALS, there is typically no identifiable family history of the disease, and its cause is believed to stem from a complex interplay of spontaneous genetic mutations and environmental factors. Familial ALS, conversely, constitutes about 10-15% of cases and indicates the presence of an inherited genetic mutation. This form often suggests a higher likelihood of the disease being passed down through generations, though its transmission is not always guaranteed.
Genetic Underpinnings: When Genes Play a Role
In familial ALS, specific genetic mutations are directly linked to the development of the disease. Scientists have successfully identified several genes that, when altered, can lead to ALS. Among the most significant is the SOD1 gene, which encodes the enzyme superoxide dismutase 1. Mutations in this gene can cause a specific form of familial ALS. Another critical gene implicated in a substantial percentage of familial cases is the C9orf72 gene. Other genes, such as the FUS genes, are also being investigated for their role. The discovery of these genes has been instrumental in deepening our understanding of disease mechanisms and has paved the way for the development of targeted therapies. For instance, Tofersen (Qalsody) is a groundbreaking therapy specifically designed for individuals with ALS caused by SOD1 gene mutations. This discovery highlights how understanding gene changes is pivotal for developing personalized treatment approaches. Comprehensive genetic testing can identify these mutations, offering crucial information for individuals and families.
Environmental Factors: The Search for External Triggers
While genetics clearly underpin familial ALS, the origins of sporadic ALS are less understood and are hypothesized to involve a complex interaction between genetic susceptibility and various environmental factors. Researchers are actively investigating a wide range of potential triggers, including exposure to certain toxins, viral infections, and even prolonged or extreme physical exertion. However, definitive links between specific environmental exposures and the subsequent development of ALS remain elusive. The ongoing search for external triggers is critically important, as identifying modifiable risk factors could unlock new avenues for prevention and treatment strategies. Emerging research also delves into the role of epigenetics—changes in gene expression that occur without altering the underlying DNA sequence—as another potential layer of complexity in ALS development.
Recognizing the Signs: A Comprehensive Look at ALS Symptoms
Early and accurate recognition of ALS symptoms is critical for timely diagnosis and intervention, though the disease’s insidious onset can make this challenging. The initial manifestations of ALS often begin subtly and can be easily mistaken for minor strains, fatigue, or everyday aches and pains.
Early Manifestations: Subtle Clues to Watch For
Common early signs of ALS include:
- Muscle weakness: This is frequently the first noticeable symptom, typically appearing in one limb or one side of the body. It might manifest as foot drop—difficulty lifting the front of the foot while walking—clumsiness, or trouble performing everyday tasks like buttoning a shirt, opening a jar, or gripping objects.
- Muscle cramps and twitches: Involuntary muscle spasms (fasciculations) and persistent cramps can be early indicators, often felt as a subtle rippling under the skin or a feeling of pins and needles.
- Speech difficulties: Slurring of words (dysarthria) or changes in vocal quality, such as a strained or nasal tone, can occur as the muscles involved in speech begin to weaken.
- Swallowing problems: Early difficulties with swallowing (dysphagia) might present as a sensation of food sticking in the throat, frequent coughing during meals, or changes in voice after eating.
Progressive Impact on Body Functions
As ALS progresses, the impact on body functions becomes more profound and widespread, affecting essential capabilities.
- Mobility: Progressive muscle weakness eventually leads to significant difficulty walking, standing, and maintaining balance. This often necessitates the use of mobility aids such as canes, walkers, or wheelchairs to maintain safety and some degree of independence.
- Speech and Communication: Severe muscle weakness can render speech unintelligible, leading to profound challenges in communication. Individuals may require augmentative and alternative communication (AAC) devices, such as speech-generating devices, to express themselves.
- Swallowing and Nutrition: As dysphagia worsens, eating and drinking can become dangerous due to an increased risk of aspiration (food or liquid entering the airways). This can lead to malnutrition, dehydration, and an increased risk of aspiration pneumonia. In many cases, a feeding tube becomes necessary to ensure adequate nutrition and hydration; this is often a gastrostomy tube, surgically placed directly into the stomach.
- Respiration: The diaphragm and other muscles essential for breathing can become progressively weaker. This can lead to respiratory failure, a life-threatening complication. Individuals may require breathing support, often starting with non-invasive ventilation such as BiPAP or CPAP machines, to assist with respiration, especially during sleep. In advanced stages, more intensive respiratory support may be needed.
Understanding ALS Subtypes and Their Presentation
While the core pathology of ALS involves the degeneration of motor neurons, the specific pattern and rate of this progression can vary significantly among individuals. This leads to different clinical presentations, often categorized into subtypes. Some individuals may experience more prominent bulbar-onset ALS symptoms, affecting speech and swallowing, very early in the disease course. Others might present with limb-onset ALS, where weakness in the arms or legs is the primary initial symptom. While the classification of ALS subtypes doesn’t alter the fundamental disease process, it can help clinicians anticipate the typical symptom progression and tailor management strategies more effectively. Other related conditions include progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), and progressive bulbar palsy (PBP), which represent variations in the affected motor neuron populations and their clinical manifestations.
Navigating Diagnosis: Identifying ALS
Diagnosing Amyotrophic Lateral Sclerosis (ALS) is a complex process that requires careful evaluation by neurologists. It is a journey that often involves a process of elimination to rule out other conditions that may mimic its symptoms. The median diagnostic delay for ALS is approximately 11 months from the onset of the first symptom, a significant period during which the disease can progress. Furthermore, research indicates disparities in diagnosis times, with Black patients experiencing delays of approximately eight months compared to White patients, highlighting an area for improved equity in care.
The Diagnostic Journey: Ruling Out Other Conditions
Because ALS symptoms can overlap significantly with those of numerous other neurological disorders, a thorough diagnostic workup is absolutely essential. Neurologists must carefully differentiate ALS from conditions such as multiple sclerosis, spinal muscular atrophy, myasthenia gravis, and various peripheral neuropathies or myopathies. Infectious diseases like HIV or Lyme disease can also present with symptoms that might initially be mistaken for ALS. This meticulous process of differentiation ensures that the correct diagnosis is made and that appropriate, timely management and supportive care can commence.
Clinical Evaluation and Neurological Examination
The diagnostic process typically begins with a detailed medical history, where the patient describes their symptoms, their onset, and their progression. This is followed by a comprehensive neurological examination. The neurologist assesses muscle strength, tone, reflexes, coordination, balance, and gait. They will specifically look for signs indicative of both upper motor neuron involvement (such as spasticity and hyperreflexia) and lower motor neuron involvement (such as muscle atrophy, fasciculations, and weakness).
Specialized Diagnostic Tests
To support the clinical diagnosis and definitively rule out other conditions, several specialized tests may be employed:
- Electromyography (EMG) and Nerve Conduction Studies (NCS): These tests are crucial for assessing the electrical activity of muscles and the speed at which nerve impulses travel along peripheral nerves. They can help identify patterns of nerve or muscle damage consistent with ALS and are invaluable in ruling out peripheral nerve disorders or primary muscle diseases.
- Magnetic Resonance Imaging (MRI): While MRI of the brain and spinal cord typically does not show specific abnormalities in early ALS, it is a critical tool. It is used to rule out other structural causes of neurological symptoms, such as brain tumors, strokes, spinal cord lesions, or nerve compression, which could mimic ALS. MRI technology has advanced significantly, allowing for more detailed imaging.
- Blood and Urine Tests: These analyses help detect metabolic abnormalities, infections, vitamin deficiencies, or other systemic conditions that could manifest with neurological symptoms.
- Cerebrospinal Fluid (CSF) Analysis: A lumbar puncture may be performed to analyze the CSF for signs of inflammation, infection, or other abnormalities that could be related to neurological disorders other than ALS.
- Muscle Biopsy: In some cases, a muscle biopsy might be performed to examine muscle tissue directly for signs of disease, helping to differentiate ALS from primary muscle disorders.
The Promise of Biomarkers: Towards Earlier and More Accurate Diagnosis
The development of reliable biomarkers for ALS is a significant and highly active area of ongoing research. Biomarkers are measurable indicators of a biological state or condition. In the context of ALS, their discovery could potentially revolutionize diagnosis by enabling earlier detection, more accurate differentiation from mimic syndromes, more precise tracking of disease progression, and better prediction of treatment response. Researchers are exploring various avenues, including analyzing specific proteins in blood or cerebrospinal fluid, identifying unique genetic signatures, and utilizing advanced neuroimaging techniques to detect subtle changes at the cellular level. The identification of such biomarkers would represent a substantial leap forward in the effective management of this complex disease.
Managing ALS: Current Treatments and Supportive Care
While there is currently no cure for Amyotrophic Lateral Sclerosis (ALS), a combination of FDA-approved medications, comprehensive supportive therapies, and a multidisciplinary care approach can significantly help slow disease progression, manage debilitating symptoms, and crucially, improve the quality of life for individuals living with this challenging condition. There is no cure for ALS now. But FDA-approved medicines, supportive therapies, and care from different specialists can slow the disease. They also help manage symptoms. This improves life quality for people with ALS.
FDA-Approved Medications: Slowing Progression and Managing Symptoms
Several medications have received FDA approval to help manage ALS and its progression:
- Riluzole: This medication is thought to work by reducing damage to motor neurons by lowering the levels of glutamate, an excitatory neurotransmitter implicated in neuronal damage. It has been shown to modestly extend survival by a few months in people with ALS and is often one of the first treatments prescribed.
- Edaravone (Radicava): Administered intravenously (or orally with RADICAVA ORS), edaravone is an antioxidant that may help reduce the damage caused by oxidative stress, a process believed to contribute to motor neuron degeneration in ALS. Clinical trials have demonstrated that it can slow the decline in physical function for some individuals.
- Tofersen (Qalsody): This groundbreaking therapy is an antisense oligonucleotide designed to target the messenger RNA (mRNA) produced by the SOD1 gene. It is specifically approved for individuals with ALS caused by a mutation in the SOD1 gene, aiming to reduce the production of the toxic SOD1 protein. Finding these genes helped us understand how the disease works. It also helped create targeted treatments like Tofersen (Qalsody). This drug is for people with ALS caused by SOD1 gene mutations.
- Relyvrio (AMX0035): This combination therapy has shown promise in clinical trials for slowing functional decline in ALS. It is designed to reduce neuronal injury and dysfunction by targeting mitochondrial and endoplasmic reticulum stress pathways.
Comprehensive Supportive Therapies and Multidisciplinary Care
Beyond medication, a holistic approach to care is paramount for individuals with ALS. This often involves a multidisciplinary team of healthcare professionals who work collaboratively to address the diverse and complex needs of patients:
- Physical Therapy: A physical therapist helps maintain muscle strength, flexibility, and mobility for as long as possible. They also provide guidance on adaptive equipment and exercises to manage fatigue and improve balance.
- Occupational Therapy: An occupational therapist assists individuals with strategies and assistive devices to perform daily living activities, such as dressing, eating, and personal hygiene, thereby enhancing independence and safety.
- Speech Therapy: A speech-language pathologist addresses difficulties with speech (dysarthria) and swallowing (dysphagia). They provide techniques for clearer communication, recommend augmentative and alternative communication (AAC) devices, and advise on strategies for safer eating and drinking to prevent aspiration.
- Nutritional Support: Dietitians work closely with patients to ensure adequate caloric intake and hydration. They often recommend modifications to food consistency, mealtime strategies, and when necessary, assist with the process of obtaining a feeding tube (like a gastrostomy tube) to maintain nutrition and hydration, especially crucial for individuals with bulbar-onset ALS.
- Respiratory Care: Pulmonologists and respiratory therapists manage breathing difficulties. This can involve prescribing breathing support devices such as non-invasive ventilation (e.g., BiPAP machines), particularly for respiratory-onset ALS, to assist with respiration, especially during sleep, and to prevent respiratory failure. They also guide decisions regarding airway clearance techniques and potential future interventions.
- Psychological and Social Support: Addressing the emotional and psychological impact of ALS is vital. Counselors, social workers, and support groups provide crucial emotional support, resources, and guidance for patients and their families. Advanced communication devices, mobility aids, and smart home technology help people stay independent and connected.
The Horizon of Hope: Latest Research Updates and Future Directions
The scientific community is actively engaged in a race against time, driven by a profound commitment to unraveling ALS’s complexities and discovering effective treatments. The global amyotrophic lateral sclerosis (ALS) market, for instance, is anticipated to grow, demonstrating increasing investment in research and treatment development.
Deepening Our Understanding of Disease Mechanisms
Researchers are moving beyond simply identifying genes to understanding the intricate cellular and molecular pathways that lead to motor neuron death. This includes investigating protein aggregation (abnormal clumps of proteins), mitochondrial dysfunction (problems with the cell’s energy production centers), oxidative stress (damage caused by unstable molecules), and neuroinflammation (inflammation within the nervous system) as key contributors to the disease process. Scientists study protein clumps, problems with mitochondria, oxidative stress, and brain inflammation. These are key parts of the disease process. A more profound understanding of these mechanisms is essential for developing precisely targeted therapies.
Advancements in Genetic Research and Gene-Targeted Therapies
The field of genetics has revolutionized ALS research. Beyond SOD1 mutations, scientists continue to identify new genes associated with ALS and are developing innovative gene-targeted therapies. Antisense oligonucleotides and RNA interference (RNAi) technologies, like Tofersen, offer the potential to silence disease-causing genes or correct their faulty products. CRISPR-based gene editing technologies also hold promise for permanently correcting genetic defects responsible for ALS.
The Promise of Novel Therapeutic Approaches
The development pipeline for ALS therapies is expanding rapidly, exploring a range of novel approaches. These include:
- Stem Cell Therapy: Research into using stem cells to replace damaged motor neurons or provide supportive factors is ongoing, though still largely in early-stage clinical trials.
- Neuroprotective Agents: New compounds are being investigated for their ability to shield motor neurons from damage and stress.
- Anti-inflammatory Strategies: Targeting inflammatory processes within the nervous system is another avenue being explored, as inflammation is known to contribute to neurodegeneration.
- Metabolic Modulators: Investigating how to optimize cellular metabolism to support motor neuron health and resilience is also a focus.
These efforts represent a significant advancement in the fight against ALS. The substantial increase in investment, reflected in the growing global market for ALS therapies, fuels this innovation and brings renewed hope for breakthroughs in treatment.
Technological Innovations for Enhanced Quality of Life
Complementing medical research, technological advancements are playing a vital role in improving the daily lives of those with ALS. Sophisticated communication devices, advanced mobility aids, and smart home technologies are empowering individuals to maintain independence and connection with their loved ones and the world. These innovations, coupled with ongoing research into treatments and cures, offer a brighter outlook for managing ALS and significantly enhancing the quality of life for all affected.
Clinical Trials: The Gateway to New Treatments
Participation in clinical trials is a critical component of advancing ALS research. These studies offer individuals with ALS the opportunity to access investigational treatments and contribute to the collective understanding of the disease. Organizations like the ALS Therapy Development Institute and leading medical centers such as the Cleveland Clinic are at the forefront of conducting these trials, exploring everything from novel drug candidates to new therapeutic modalities. Staying informed about available clinical trials is crucial for patients and their families seeking the latest potential treatments.
Conclusion
Amyotrophic Lateral Sclerosis (ALS) presents a formidable challenge, touching the lives of thousands with its relentless progression and devastating impact. Yet, the landscape of ALS research is one of dynamic progress and unwavering hope. From deciphering the intricate roles of genes like the SOD1 gene and C9orf72 gene, and understanding complex cellular mechanisms, to developing targeted therapies such as RADICAVA ORS, and leveraging cutting-edge technologies, the scientific community is making tangible strides. The journey from initial muscle twitching and muscle weakness to diagnosis, while often fraught with delay, is becoming more informed by advancements in diagnostic tools and the pursuit of vital biomarkers. While current treatments like Riluzole, Edaravone, and Tofersen offer ways to slow progression and manage symptoms, they underscore the urgent need for further discoveries, particularly concerning the underlying causes related to the spinal cord and motor neurons.
The fight against this motor neuron disease, also known as Lou Gehrig’s disease or Charcot’s disease, is a global effort. Significant investments in research, fueled by dedicated organizations and passionate patient communities, are driving innovation in gene therapies, neuroprotection, and technological aids. While conditions like frontotemporal dementia can co-occur, adding layers of complexity, the overarching goal remains clear: to find a cure and improve the lives of those affected by ALS. Ongoing clinical trials are the crucial bridge to future treatments, offering hope and contributing vital data. Supporting research, advocating for patients, and embracing new technologies brings us closer to a future where ALS is no longer a death sentence but a treatable or even preventable condition, significantly improving outcomes and quality of life for people with ALS.
Leave a Reply