Huntington’s Disease Latest Research, Biomarker Discoveries, and Promising Treatments
A New Era of Hope for Huntington’s Disease
Huntington’s Disease (HD) has long represented a formidable challenge in neurology, a devastating inherited neurodegenerative disorder with no cure and limited symptomatic treatments. However, a wave of scientific innovation is fundamentally altering this landscape. We are entering a new era of understanding and intervention, driven by groundbreaking research into the disease’s genetic roots, the identification of critical biomarkers, and the development of novel therapeutic strategies. This confluence of progress offers unprecedented hope for patients and families grappling with this progressive condition.
Deciphering Huntington’s: A Progressive Neurodegenerative Challenge
Huntington’s Disease is a devastating inherited disorder affecting approximately 3-7 individuals per 100,000 worldwide. It is characterized by a progressive breakdown of nerve cells in the brain, leading to a triad of symptoms: motor disturbances, cognitive decline, and psychiatric complications. The disease typically manifests in adulthood, often between the ages of 30 and 50, though juvenile and late-onset forms exist. Its insidious progression means that by the time symptoms become apparent, significant neuronal damage has already occurred, making early diagnosis and intervention critical. The unpredictable nature of symptom onset and severity, coupled with the absence of disease-modifying treatments, places immense burdens on affected individuals and their families, underscoring the urgent need for scientific breakthroughs.
The urgency for breakthroughs in Huntington’s Disease research cannot be overstated. For decades, the medical community has primarily relied on symptomatic treatment to manage the debilitating effects of HD, offering little to alter its inexorable course. The emotional, physical, and financial toll on patients and their caregivers is immense. Recent advancements, however, have shifted the paradigm from mere symptom management to actively targeting the disease’s root causes. The promise of understanding the biological processes underlying HD, coupled with the development of sensitive biomarkers and innovative therapeutic strategies, has ignited a renewed sense of optimism. This convergence of scientific endeavor is vital, offering a realistic pathway towards slowing, stopping, or even reversing the disease’s progression, thereby transforming the outlook for every patient diagnosed.
Understanding Huntington’s Disease: The Genetic Imperative
At the heart of Huntington’s Disease lies a specific genetic defect. This inherited condition is caused by a mutation in the HTT gene, located on chromosome 4. This gene provides instructions for making a protein called huntingtin. The hallmark of the HD mutation is an abnormal expansion of a CAG trinucleotide repeat sequence within the HTT gene.
Typically, individuals have fewer than 26 CAG repeats. In HD, this sequence is expanded, with individuals having 40 or more CAG repeats almost certainly developing the disease. The number of repeats influences the age of onset and severity; more repeats generally lead to an earlier onset and more rapid progression. This genetic imperative is the foundational understanding upon which all subsequent research and therapeutic strategies are built, making genetic testing a crucial component of diagnosis for at-risk individuals.
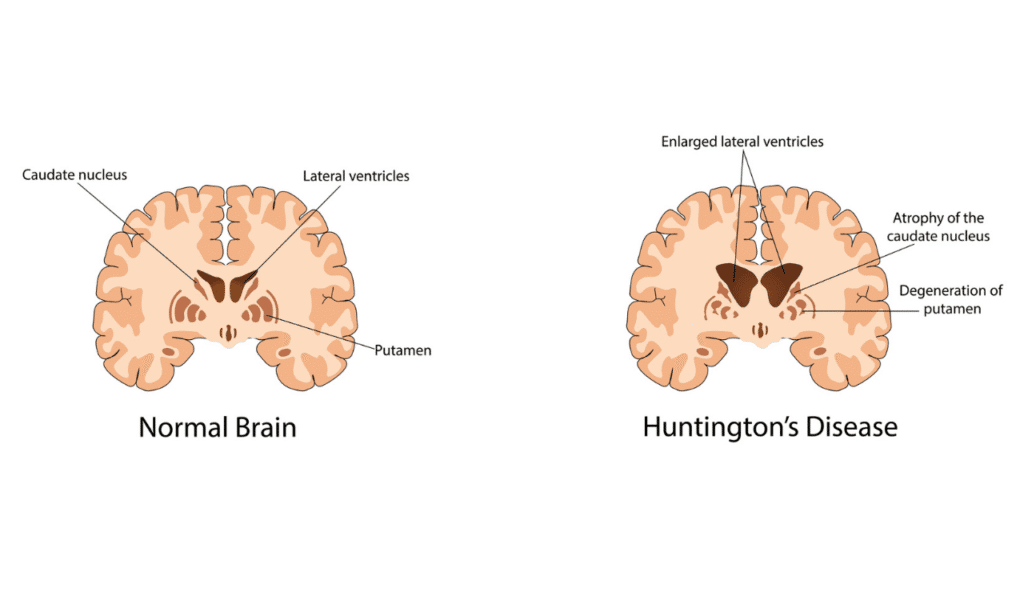
The expanded CAG repeats lead to the production of a dysfunctional and toxic form of the huntingtin protein, known as mutant huntingtin protein (mHTT). This aberrant protein behaves differently from its normal counterpart. It misfolds, aggregates, and accumulates within neurons, particularly in specific areas of the brain like the basal ganglia and cerebral cortex, which are critical for movement control, cognition, and emotional regulation.
The accumulation of mHTT triggers a cascade of detrimental biological processes, including mitochondrial dysfunction, impaired axonal transport, excitotoxicity, and inflammation, ultimately leading to neuronal death. This neurodegeneration is the direct cause of the progressive motor, cognitive, and psychiatric symptoms experienced by the patient.
Current interventions primarily focus on managing these symptoms, such as using antipsychotics or antidepressants to address behavioral issues, or medications like tetrabenazine to control involuntary movements (chorea). However, these treatments do not halt or reverse the underlying neuronal damage, highlighting a significant unmet need for disease-modifying therapies. The doctor plays a pivotal role in managing these symptoms and guiding the patient through the complex care landscape.
Advanced Biomarker Discoveries in HD
Biomarkers are objective, measurable indicators that reveal the presence of a biological state, condition, or process. In the context of Huntington’s Disease, biomarkers are indispensable tools for diagnosis, understanding disease progression, and evaluating the effectiveness of potential treatments. They allow researchers and clinicians to peer inside the complex biological processes occurring within the brain and body, even before overt symptoms manifest or when symptoms are subtle. The discovery and validation of robust biomarkers are fundamental to accelerating drug development and improving patient care, offering a more precise and sensitive way to monitor the disease’s trajectory than solely relying on clinical observations.
One of the most significant advancements in biomarker discovery for neurodegenerative diseases, including HD, is the identification of neurofilament light chain (NfL). NfL is a structural protein found in neurons. When neurons are damaged or degenerate, their components, including NfL, are released into the extracellular space and subsequently detectable in bodily fluids. Elevated levels of NfL in the blood or cerebrospinal fluid (CSF) serve as a sensitive indicator of ongoing neuronal injury. This has profound implications for HD: NfL levels can reflect disease severity, predict the rate of progression, and are often elevated even in preclinical stages. The ability to measure neuronal damage via a simple blood draw is a game-changer for diagnosis, prognosis, and for tracking the impact of therapeutic interventions in clinical trials, offering a valuable surrogate endpoint.
The mutant huntingtin protein (mHTT) itself stands as a critical and direct biomarker in Huntington’s Disease. Unlike NfL, which indicates neuronal damage, mHTT is the molecular culprit. Detecting the presence of mHTT, especially in its toxic aggregated forms, directly confirms the disease pathology. Advances in assay technologies have enabled the measurement of mHTT in biological fluids. For instance, detecting levels of mHTT in cerebrospinal fluid provides direct evidence of the disease process in the brain. Researchers are also exploring ways to measure mHTT and its downstream effects in the blood, which would offer an even more accessible method for monitoring disease activity and treatment response. Understanding and quantifying mHTT is paramount for developing and testing therapies designed to reduce its levels or mitigate its toxic effects.
Beyond NfL and mHTT, a diverse array of other protein biomarkers and their altered forms (proteoforms) are being investigated. These include proteins involved in cellular stress responses, protein degradation pathways, inflammation, and synaptic function, all of which are implicated in HD pathogenesis. Mass spectrometry and advanced proteomic techniques are instrumental in identifying these subtle molecular changes. These biomarkers may offer a more nuanced understanding of specific biological processes affected in HD and could potentially serve as prognostic biomarkers, predicting disease trajectory, or as predictive biomarkers, indicating how a patient might respond to a particular therapeutic strategy. For instance, changes in levels of certain chaperones or ubiquitin ligases could signal cellular attempts to cope with or clear toxic proteins, providing insights into disease mechanisms.
Genetic and molecular biomarkers are also crucial for a comprehensive understanding of Huntington’s Disease. While the primary genetic mutation (CAG repeat expansion) is the diagnostic cornerstone, other genetic and RNA-based markers are being explored. These can include variations in gene expression profiles or the presence of specific RNA species that correlate with disease severity or progression. Analyzing DNA and RNA from patient samples can offer deeper insights into the complex molecular machinery affected by the HTT mutation and may reveal novel targets for intervention or serve as predictive biomarkers for treatment efficacy. These molecular insights are vital for unraveling the intricate biological processes that contribute to neuronal dysfunction.
The advent of digital biomarkers and wearable technology is ushering in a new frontier in HD research. These tools capture continuous, objective data about a patient’s functional status, such as gait patterns, tremor, sleep disturbances, and activity levels, using sensors in smartphones or wearable devices. These digital markers can provide a real-time, longitudinal assessment of disease progression and treatment response, complementing traditional clinical assessments. They have the potential to offer subtle insights into motor and cognitive function that might be missed during infrequent doctor visits and can serve as highly sensitive tools for detecting changes in biological processes indicative of disease modification.
Despite the exciting progress, significant challenges remain in the journey to clinical validation for HD biomarkers. Ensuring reliability, reproducibility, and standardization across different laboratories and patient populations is paramount. Biomarkers must undergo rigorous testing to prove their accuracy, sensitivity, and specificity. Establishing them as definitive diagnostic biomarkers, reliable prognostic biomarkers, or predictive biomarkers that can guide clinical decision-making requires extensive validation through large-scale clinical trials. Furthermore, regulatory approval is necessary for these biomarkers to be integrated into routine clinical practice, ensuring that every patient benefits from these advancements.
Promising Therapeutic Strategies in Development
The landscape of Huntington’s Disease treatment is undergoing a profound transformation, moving beyond symptom management to target the underlying genetic and molecular causes of the disease.
A variety of innovative therapeutic strategies are in various stages of drug development, offering unprecedented hope for altering the course of this devastating illness. These interventions aim to address the root of the problem by tackling the toxic mutant huntingtin protein (mHTT) or by modulating the biological processes that lead to neuronal damage.
A leading approach in HD therapeutic development focuses on gene silencing and gene editing technologies. Gene silencing strategies, such as antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs), are designed to reduce the production of mHTT. These molecules work by targeting the messenger RNA (mRNA) transcribed from the mutated HTT gene, effectively instructing the cell to degrade the faulty mRNA before it can be translated into the toxic protein.
Several ASO-based therapies are currently in clinical trials, demonstrating the potential to lower mHTT levels in the brain. Gene editing technologies, like CRISPR-Cas9, represent a more futuristic but potentially powerful intervention, aiming to directly correct the genetic mutation in the HTT gene itself, thereby preventing the production of mHTT altogether. While gene editing for HD is still in its early stages of preclinical research, it holds immense promise for a permanent genetic intervention.
Beyond directly targeting the genes or their transcripts, researchers are exploring various strategies to modulate disease pathways and combat the toxic effects of mHTT. This includes developing small molecules designed to interfere with the aggregation of mHTT, enhance cellular mechanisms for clearing misfolded proteins (like autophagy), or protect neurons from excitotoxicity and oxidative stress.
Another area of interest is targeting specific protein interactions that contribute to mHTT toxicity. Furthermore, drawing parallels from advancements in cancer research, particularly in areas like targeted protein degradation (TPD), offers exciting avenues.
TPD strategies can harness the cell’s own machinery to specifically tag and destroy harmful proteins, like mHTT. This approach, proven effective against certain cancer proteins, is being adapted to clear toxic huntingtin and represents a novel intervention strategy.
The effectiveness of these cutting-edge therapeutic strategies is being rigorously evaluated in clinical trials. These trials are essential for determining the safety and efficacy of new drugs and interventions in human patients. The design of these trials is increasingly sophisticated, often incorporating a range of biomarkers to measure treatment response. For instance, trials may monitor changes in NfL levels as an indicator of reduced neuronal damage or track reductions in mHTT levels in CSF or blood as a direct measure of drug activity. The patient’s experience, including motor, cognitive, and psychiatric assessments, remains central to evaluating overall benefit, but biomarkers provide crucial objective data.
Accelerating Clinical Research and Validation
The journey from a scientific discovery in the laboratory to an approved treatment available to patients is long and arduous, particularly for complex neurodegenerative diseases like Huntington’s. The rigorous process of drug development involves multiple phases of preclinical testing followed by sequential clinical trials designed to establish safety and efficacy. For HD, this process is being accelerated through strategic approaches that leverage our growing understanding of the disease’s biological processes and the power of biomarkers. The development strategy often involves close collaboration between academic researchers, pharmaceutical companies, and patient advocacy groups.
A critical component in expediting drug development is the integration of biomarkers as key endpoints in clinical trials. Traditionally, clinical trials relied on observing changes in clinical symptoms, which can take years to manifest or change significantly. However, by using biomarkers, researchers can potentially detect treatment effects much earlier and more objectively. Diagnostic biomarkers help in selecting appropriate patient populations, while prognostic biomarkers can help stratify patients based on expected disease progression. Predictive biomarkers can identify individuals most likely to respond to a specific treatment.
Furthermore, measuring changes in biomarkers like NfL or mHTT can serve as surrogate endpoints, allowing for a faster assessment of a drug’s potential effectiveness, thereby speeding up the overall drug development timeline.
Global collaboration and data sharing are paramount to accelerating Huntington’s Disease research and advancing therapeutic strategies. No single institution or country can tackle such a complex disease alone.
Establishing unified research strategies, sharing data openly, and pooling resources across international consortia are vital. This collaborative approach ensures that research efforts are not duplicated, that promising leads are pursued efficiently, and that diverse patient populations are included in studies. This collective strategy helps in rapidly validating biomarkers and testing potential treatments in large, well-characterized cohorts, ultimately benefiting every patient worldwide.
Ethical Considerations and Patient-Centered Impact
Navigating the landscape of Huntington’s Disease research and treatment involves significant ethical considerations, with the patient’s well-being and autonomy at the forefront. For individuals at risk, the option of predictive testing for the genetic mutation presents a complex ethical decision.
This process requires comprehensive genetic counseling to ensure that individuals fully understand the implications of a positive result, including the lack of a cure and the potential impact on family members. The doctor’s role in providing empathetic and thorough guidance is crucial, empowering individuals to make informed choices about their genetic destiny and to plan for their future health and financial needs.
The advent of powerful gene-editing technologies, while holding immense promise for inherited diseases, also brings forth profound ethical implications. Questions surrounding germline editing (changes that can be passed to future generations), off-target effects, and equitable access to these advanced therapies must be carefully considered.
As research progresses towards interventions that can fundamentally alter the genetic makeup of an individual, robust ethical frameworks and public discourse are essential to guide responsible innovation. The impact on future generations and the potential for unintended consequences require meticulous scientific scrutiny and societal consensus.
Ultimately, the success of any breakthrough in Huntington’s Disease research hinges on its positive impact on the patient and their family. Empowering patients with access to accurate information, comprehensive support services, and opportunities for advocacy is critical. Patient advocacy groups play a vital role in driving research, raising awareness, and ensuring that patient perspectives are integrated into every stage of drug development and clinical care.
A patient-centered strategy ensures that treatments are not only scientifically sound but also address the real-world needs and challenges faced by individuals living with HD, improving their quality of life and fostering a sense of hope and agency.
The Road Ahead: Future Directions in Huntington’s Disease Research
The future of Huntington’s Disease research is brimming with potential, fueled by an ever-deepening understanding of the disease’s intricate biological processes and the relentless pursuit of effective interventions. The ongoing revolution in our ability to decipher complex biological systems and to develop targeted therapies promises to transform the lives of patients and their families. The focus is shifting towards more personalized and precise approaches, moving beyond broad interventions to tailor treatments to individual needs and disease profiles.
Advancing through multi-omics and systems biology approaches represents a significant leap forward. By integrating data from genomics, transcriptomics, proteomics, and metabolomics, researchers can gain a holistic view of the complex interactions within cells and biological systems affected by HD.
This comprehensive analysis allows for the identification of novel therapeutic targets, the development of more accurate biomarkers, and a deeper understanding of the disease’s heterogeneity. Systems biology offers a powerful lens through which to unravel the intricate cascade of events initiated by the HTT gene mutation, paving the way for more sophisticated and effective intervention strategies. The ultimate goal is to leverage these integrated insights to develop personalized medicine approaches, ensuring that each patient receives the most effective treatment strategy for their unique biological profile, thereby ushering in an era of transformed care for Huntington’s Disease.
A Future Transformed by HD Breakthroughs
The landscape of Huntington’s Disease research is rapidly evolving, marking a significant shift from decades of limited options to a future characterized by unprecedented hope and tangible progress. Groundbreaking discoveries in understanding the genetic imperative, the identification of sophisticated biomarkers, and the development of innovative therapeutic strategies are collectively reshaping the fight against this devastating neurodegenerative disorder. The journey has been arduous, but the convergence of scientific efforts, particularly in unraveling the role of the HTT gene and the toxic huntingtin protein, has laid a robust foundation for change.
The emergence of advanced biomarkers, especially those detectable in blood like Neurofilament Light Chain (NfL) and direct measurements of mutant huntingtin protein (mHTT), is revolutionizing diagnosis, enabling earlier detection, and providing objective measures for tracking disease progression and treatment efficacy. These biological markers are instrumental in accelerating clinical trials, allowing for more precise evaluation of therapeutic interventions. Alongside these diagnostic advancements, a diverse array of promising treatments are in development. Gene silencing and gene editing technologies, alongside strategies targeting protein degradation and modulating disease pathways, offer the potential to address the root causes of HD rather than just managing symptoms.
The rigorous path of drug development, underscored by ongoing clinical trials, is being significantly enhanced by the strategic use of biomarkers as endpoints. Global collaboration and data sharing are critical to this acceleration, fostering a unified strategy that benefits the entire patient community. Crucially, ethical considerations and a steadfast focus on the patient experience are paramount, ensuring that scientific progress translates into meaningful improvements in quality of life and support for individuals and their families.
Looking ahead, the integration of multi-omics and systems biology promises an even deeper, more holistic understanding of HD’s complex biological processes, paving the way for truly personalized medicine. While challenges remain, the current trajectory of research and development offers a powerful testament to scientific ingenuity and collective dedication. This is a pivotal moment, offering a renewed vision of a future where Huntington’s Disease is not only better understood and managed but potentially treatable, or even preventable, transforming the lives of countless patients worldwide.


Leave a Reply