Frontotemporal Dementia
Frontotemporal Dementia (FTD) is a group of progressive brain disorders that primarily affect the frontal and temporal lobes of the brain. Unlike the more commonly known Alzheimer’s disease, FTD often emerges at a younger age, typically between 45 and 65, and its initial symptoms are often characterized by profound changes in personality, behavior, and language rather than memory loss. This insidious onset and distinct symptom profile can make FTD challenging to diagnose, often leading to initial misdiagnoses as psychiatric conditions or other forms of dementia. Understanding the nuances of FTD is crucial for early identification, appropriate management, and providing effective support to individuals and their families navigating this complex neurological condition. This article aims to provide a comprehensive overview of FTD, exploring its unique characteristics, the underlying causes, the diagnostic process, and the available approaches to management and support.
Introduction to Frontotemporal Dementia
Frontotemporal Dementia (FTD) represents a spectrum of neurodegenerative diseases that are often misunderstood and underdiagnosed. These conditions share a common pathology involving the degeneration of the frontal and temporal lobes, brain regions critical for regulating personality, behavior, language, and executive functions. The progressive nature of FTD means that symptoms worsen over time, leading to significant disability and impacting all aspects of a person’s life.
What is Frontotemporal Dementia (FTD)?
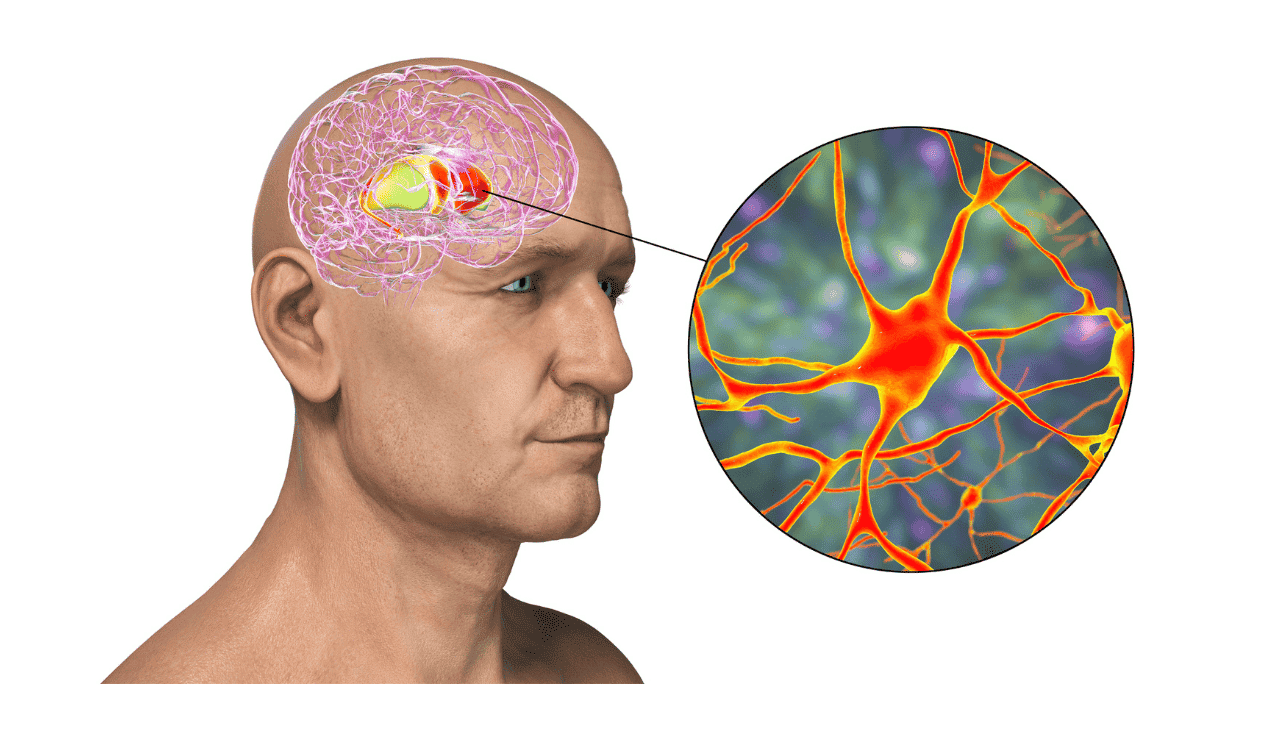
![]() Key differences between Frontotemporal Dementia (FTD) and Alzheimer’s disease, highlighting the distinct brain regions, age of onset, and initial symptoms.
Key differences between Frontotemporal Dementia (FTD) and Alzheimer’s disease, highlighting the distinct brain regions, age of onset, and initial symptoms.
Frontotemporal Dementia (FTD) is an umbrella term for a group of progressive brain disorders caused by degeneration of the frontal and temporal lobes. These lobes are vital for our personality, behavior, judgment, reasoning, and language abilities. Unlike Alzheimer’s disease, which often begins with memory problems, FTD typically first affects behavior, personality, or language skills. It is a significant cause of dementia in individuals under the age of 65, highlighting its impact on a relatively younger demographic compared to other dementia types. The underlying pathology involves the gradual death of nerve cells in these specific areas of the brain.
Why Understanding FTD is Crucial
Understanding FTD is paramount due to its distinct presentation and the profound impact it has on individuals and their families. Its early onset means it frequently affects people during their peak working years, disrupting careers, family dynamics, and social engagement. The significant changes in personality and behavior, or difficulties with language, can be distressing and bewildering for both the individual and their loved ones. Early and accurate diagnosis is crucial for initiating appropriate therapy and support, managing symptoms effectively, and making informed decisions about future care. Recognizing FTD’s unique characteristics helps differentiate it from other forms of dementia, leading to more targeted interventions and a better quality of life for those affected.
FTD vs. Other Dementias: A Unique Neurodegenerative Disorder
Frontotemporal Dementia (FTD) stands apart from other common forms of dementia, particularly Alzheimer’s disease, due to its age of onset, affected brain regions, and primary symptom profile. Understanding these distinctions is key to accurate diagnosis and management.
Distinguishing FTD from Alzheimer’s Disease
Alzheimer’s disease is the most common cause of dementia, typically affecting older adults and primarily characterized by memory loss. In contrast, Frontotemporal Dementia (FTD) often strikes individuals earlier, usually between the ages of 45 and 65. While memory can be affected in later stages of FTD, the initial hallmarks are typically changes in behavior, personality, or language. For instance, a person with FTD might exhibit disinhibition or loss of empathy, whereas someone with early Alzheimer’s is more likely to forget recent events. This difference in primary symptoms is a critical diagnostic clue.
The Brain Regions Affected: Frontal and Temporal Lobes
The hallmark of FTD is the progressive degeneration of the frontal and temporal lobes of the brain. The frontal lobes are the command center for executive functions, including decision-making, impulse control, social behavior, and personality. Damage here can lead to significant behavioral changes. The temporal lobes are crucial for understanding language, processing emotions, and memory formation. When these lobes are affected, individuals may experience profound language deficits, known as aphasia, or difficulties with social cognition and emotional understanding. This localized brain damage is the root cause of the specific symptoms seen in FTD.
The Diverse Faces of FTD: Types and Their Characteristic Symptoms
FTD is not a single entity but rather a group of disorders that manifest in various ways, largely depending on which part of the frontal and temporal lobes are most affected. These different forms present with distinct constellations of symptoms, making it a complex condition to navigate.
Behavioral Variant Frontotemporal Dementia (bvFTD)
Behavioral Variant Frontotemporal Dementia (bvFTD) is the most common subtype of FTD. It primarily impacts the frontal lobes, leading to prominent changes in behavior and personality. Individuals with bvFTD often experience a loss of social graces, exhibiting disinhibition, impulsivity, or inappropriate social conduct. They may show a decline in empathy, become apathetic, or develop rigid routines and compulsive behaviors. Changes in eating habits, such as overeating or developing new cravings, are also common. These altered behaviors can be mistaken for psychiatric disorders, complicating early diagnosis.
Primary Progressive Aphasia (PPA): Language-Led Dementia
Primary Progressive Aphasia (PPA) is a form of FTD where the initial and most prominent symptoms involve language impairment. PPA is characterized by progressive difficulties with speech and language, distinct from the memory loss typically seen in Alzheimer’s disease. Several variants of PPA exist, each affecting different aspects of language. One key variant is semantic dementia, also known as Semantic Variant PPA (svPPA), where individuals lose the ability to understand the meaning of words and objects, struggling with word retrieval and naming. Another common form is Progressive Nonfluent Aphasia (PNFA) or Logopenic Variant PPA (lvPPA), which affects speech production, leading to halting speech, grammatical errors, and word-finding difficulties. PPA directly impacts communication, a fundamental aspect of daily life.
FTD with Motor Neuron Disease (FTD-ALS)
A significant overlap exists between FTD and Amyotrophic Lateral Sclerosis (ALS), also known as motor neuron disease. In approximately 10-20% of ALS cases, individuals develop behavioral or cognitive changes characteristic of FTD. Conversely, some individuals with FTD may later develop motor neuron symptoms. This combined presentation, known as FTD-ALS, involves both the degeneration of brain areas controlling behavior and language, and the degeneration of motor neurons that control voluntary muscle movement. Symptoms can include muscle weakness, spasticity, and speech difficulties alongside the characteristic behavioral or language changes of FTD.
Related Syndromes within the FTD Spectrum
The Frontotemporal Dementia (FTD) spectrum encompasses several other related neurodegenerative disorders that share neuropathological features and can present with overlapping symptoms. While distinct conditions, they share a common pathway of degeneration in the frontal and temporal lobes, or related cortical areas. Among these are Progressive Supranuclear Palsy (PSP), which primarily affects balance, eye movements, and swallowing, and Corticobasal Degeneration (CBD), which can cause limb apraxia, rigidity, and sensory deficits. Historically, Pick’s disease was a recognized entity within FTD, characterized by the presence of specific tau protein aggregates (Pick bodies) and severe atrophy, particularly in the temporal lobes. Understanding these related syndromes is important for differential diagnosis.
Decoding the Causes: What Happens in the Brain?
The underlying causes of Frontotemporal Dementia (FTD) are complex and involve a combination of genetic predispositions and the accumulation of abnormal proteins within the brain. This leads to progressive damage and loss of nerve cells in the affected frontal and temporal lobes.
Brain Atrophy and Degeneration in Frontal and Temporal Lobes
At the core of FTD is the progressive degeneration of neurons, particularly in the frontal and temporal lobes. This neuronal death leads to significant shrinkage, or atrophy, of these brain regions. The loss of brain tissue directly correlates with the decline in cognitive, behavioral, and language functions associated with FTD. The specific patterns of atrophy can often help distinguish FTD from other neurodegenerative conditions and even between different subtypes of FTD, guiding the diagnosis. This brain damage is not sudden but a gradual process that unfolds over years.
The Role of Abnormal Proteins: Tau and TDP-43
A key pathological feature in many FTD cases is the abnormal accumulation of specific proteins within brain cells. The two most implicated proteins are tau and TDP-43. In some forms of FTD, such as Pick’s disease, tau proteins misfold and aggregate into abnormal tangles. In other, more common forms, TDP-43, a protein normally involved in gene expression, also misfolds and forms toxic inclusions within neurons. The accumulation of these abnormal proteins disrupts normal cellular function, leading to neuronal stress, death, and subsequent degeneration of the affected brain areas. These proteinopathies are central to understanding the pathogenesis of FTD.
Genetic Factors and Familial FTD
While most cases of FTD are sporadic, a significant proportion, estimated around 30-40%, have a genetic component, leading to familial FTD. This form often follows an autosomal dominant inheritance pattern, meaning a single copy of a mutated gene is sufficient to cause the disease. Several genes have been identified as culprits, including C9orf72 (the most common genetic cause), GRN (progranulin), and MAPT (which codes for tau protein). Genetic testing can be recommended for individuals with a strong family history of FTD or early-onset dementia. However, for the majority of FTD cases, the exact genetic or environmental triggers remain unclear, making the condition more challenging to predict or prevent.
The Diagnostic Journey: How FTD is Identified
Diagnosing Frontotemporal Dementia (FTD) can be a complex and lengthy process due to the varied nature of its symptoms and its potential to mimic other neurological and psychiatric conditions. A comprehensive approach involving multiple assessments is typically required.
Challenges in Diagnosing FTD
The primary challenge in diagnosing FTD lies in its diverse presentation. The prominent changes in behavior and personality in bvFTD can easily be mistaken for psychiatric disorders like depression, bipolar disorder, or frontotemporal lobe epilepsy. Similarly, Primary Progressive Aphasia (PPA) can be misdiagnosed as a stroke or primary language disorder. Because memory loss is not usually an early symptom, it is often overlooked by clinicians and families as a potential sign of dementia, especially in younger individuals. This diagnostic ambiguity means that many individuals with FTD may undergo years of misdiagnosis and inappropriate treatment before the correct diagnosis is established.
Comprehensive Clinical Evaluation and Medical History
A thorough clinical evaluation and detailed medical history are foundational to diagnosing FTD. This involves gathering information not only from the patient but also from family members or close companions who can provide crucial insights into changes in behavior, personality, language, and daily functioning. The clinician will assess cognitive abilities, neurological function, and observe for specific behavioral patterns. Understanding the onset and progression of symptoms is critical for differentiating FTD from other conditions.
Cognitive and Neuropsychological Testing
Cognitive and neuropsychological testing plays a vital role in assessing the specific deficits associated with FTD. These tests evaluate various domains, including executive functions (planning, problem-solving, impulse control), language abilities (fluency, comprehension, word retrieval), social cognition, and attention. Standardized tests can identify subtle impairments in these areas, which are often affected early in FTD, even when memory remains relatively intact. This detailed assessment helps pinpoint the pattern of cognitive decline characteristic of FTD and distinguish it from other forms of dementia.
Advanced Brain Imaging Techniques
Advanced brain imaging techniques, such as Magnetic Resonance Imaging (MRI) and Positron Emission Tomography (PET) scans, are invaluable tools in the diagnosis of FTD. MRI can reveal patterns of atrophy, showing shrinkage in the frontal and temporal lobes that is characteristic of FTD. Different subtypes of FTD may show distinct patterns of volume loss. PET scans can measure brain metabolism or the presence of specific proteins like amyloid and tau, helping to differentiate FTD from Alzheimer’s disease and other neurodegenerative conditions. These imaging techniques provide objective evidence of the underlying neuropathology.
Cerebrospinal Fluid (CSF) Analysis and Lumbar Puncture
In some cases, cerebrospinal fluid (CSF) analysis may be employed as part of the diagnostic process. A lumbar puncture (spinal tap) is performed to collect CSF, which is then analyzed for specific biomarkers. While not as definitive for FTD as it is for some other neurological conditions, CSF analysis can help rule out infections or inflammatory conditions and may reveal certain protein profiles or genetic markers that support an FTD diagnosis, particularly in differentiating it from Alzheimer’s disease.
Genetic Testing: When and Why it’s Recommended
Genetic testing is typically considered for individuals with a strong family history of FTD or early-onset dementia, as it can confirm a diagnosis of familial FTD. If a specific gene mutation (like C9orf72, GRN, or MAPT) is identified, it can definitively diagnose a genetic form of FTD. This information can be crucial for other family members who may be at risk and for reproductive counseling. However, genetic testing is not typically performed for sporadic cases where no family history is present, as it does not alter immediate treatment strategies but may have significant implications for genetic counseling and family planning.
Establishing a Diagnosis: Current Diagnostic Criteria
The diagnosis of FTD is established based on a combination of clinical findings, neurological examination, cognitive and neuropsychological testing, brain imaging results, and sometimes genetic testing or CSF analysis. Clinicians rely on established diagnostic criteria, such as those developed by the International Consensus Project on Clinical Diagnostic Criteria for FTD. These criteria help guide the diagnostic process, ensuring that FTD is accurately identified and differentiated from other conditions. Definitive diagnosis often requires experienced specialists in neurology, neuropsychology, and behavioral neurology.
Managing FTD: Treatment, Care, and Support
While there is currently no cure for Frontotemporal Dementia (FTD), a multidisciplinary approach focusing on managing symptoms, providing robust support, and improving quality of life is essential for individuals affected by this progressive disease and their families.
Current Treatment Approaches: Focusing on Symptom Management
Treatment for FTD is primarily focused on managing the wide range of symptoms. For behavioral changes, therapy and environmental modifications are often employed. Medications may be prescribed to manage agitation, depression, or anxiety, although their effectiveness can vary. For language difficulties associated with PPA, speech and language therapy can help individuals maintain communication skills for as long as possible. Physical therapy and occupational therapy can assist with motor symptoms and daily living activities. While there are no disease-modifying drugs for FTD, research is ongoing into potential treatments that target the underlying proteinopathies.
Practical Support for Caregivers and Families
Caregiving for someone with FTD is often demanding, as the behavioral and personality changes can be particularly challenging to manage. Practical support for caregivers is crucial. This includes education about the disease, strategies for managing challenging behaviors, assistance with financial and legal planning, and respite care. Emotional support groups and counseling can help caregivers cope with the stress and grief associated with FTD. Understanding the progressive nature of the condition and planning for future care needs is an essential part of the support process.
Finding Community and Resources
Connecting with support organizations and communities can provide invaluable resources for individuals with FTD, their families, and caregivers. Organizations like The Association for Frontotemporal Degeneration (AFTD) and the Alzheimer’s Association offer information, support networks, and advocacy. These groups provide a platform for sharing experiences, accessing practical advice, and staying informed about the latest research and treatment advancements. Building a strong support system is vital for navigating the challenges of living with FTD.
The Outlook for FTD and Future Directions in Research
The prognosis for Frontotemporal Dementia (FTD) is generally characterized by progressive decline. The rate of progression varies significantly among individuals and depends on the specific subtype of FTD and the underlying pathology. While current management focuses on symptom relief and support, the future holds promise through ongoing research efforts aimed at understanding the disease’s fundamental mechanisms and developing effective treatments.
Researchers are actively investigating novel therapeutic targets, including those that aim to clear abnormal proteins like tau and TDP-43, and gene therapy approaches for genetic forms of FTD. Advances in brain imaging and biomarkers are improving the accuracy and speed of diagnosis, which is critical for timely intervention. The hope is that a deeper understanding of the degeneration process in the frontal and temporal lobes will lead to therapies that can slow, halt, or even reverse the course of FTD, offering a brighter outlook for those affected.
Conclusion: Living with FTD and the Path Forward
Frontotemporal Dementia (FTD) presents a unique set of challenges within the broader landscape of dementia. Its impact on personality, behavior, and language, often striking at younger ages, necessitates a distinct understanding and approach compared to conditions like Alzheimer’s disease. The degeneration of the frontal and temporal lobes, driven by complex factors including abnormal proteins and genetics, underscores the intricate nature of this neurological disorder.
The journey from recognizing initial symptoms to receiving an accurate diagnosis can be arduous, fraught with challenges due to FTD’s varied presentations. However, through comprehensive clinical evaluation, advanced brain imaging, and dedicated neuropsychological testing, clinicians are increasingly adept at identifying this condition. While a cure remains elusive, effective management strategies, including targeted therapy and robust support systems for individuals and their families, can significantly improve quality of life. Continued research into the underlying causes, particularly the roles of tau and TDP-43 proteins, and the development of innovative treatments, offers hope for a future where FTD can be more effectively treated, managed, and perhaps even prevented. Navigating FTD requires resilience, informed support, and a commitment to advancing our collective understanding of this complex disease.


Leave a Reply