
Why CSF Biomarkers Break Down in Late-Stage CNS Trials
CSF biomarkers aren’t failing because the biology is wrong. They’re failing because lumbar puncture is incompatible with late-stage trial demands. Here’s what breaks down — and what’s replacing it.
Key Takeaways
- Lumbar puncture limits patients to 2–3 CSF timepoints per trial, making dense PK/PD sampling operationally impossible
- CNS biomarker specificity in CSF erodes in late-stage disease, when peripheral confounders and blood-brain barrier disruption degrade the signal
- LP-related patient dropout of 10–20% in long trials systematically biases the biomarker-selected population
- Neuron-derived extracellular vesicles (NDEVs) isolated from blood preserve CNS specificity while enabling unlimited sampling timepoints
- L1CAM-directed NDEV enrichment provides a practical path to dense, longitudinal CNS pharmacodynamic monitoring without spinal access
The Problem Isn’t the Biology
When a Phase II CNS trial fails to show pharmacodynamic signal, the reflex is to interrogate the drug. Was the dose wrong? Was the target engaged? Did the compound even reach the brain?
These are fair questions. But increasingly, the evidence points elsewhere. The biomarker strategy — specifically, the decision to rely on cerebrospinal fluid (CSF) collected by lumbar puncture — is often the variable that breaks first.
This isn’t a controversial claim. Lumbar puncture has been the gold standard for CNS biomarker collection for decades, and for good reason: CSF bathes the brain and spinal cord directly, and proteins shed by neurons and glia appear in CSF at concentrations that reflect CNS pathology with reasonable fidelity. In early-phase, proof-of-concept studies with small patient cohorts and minimal sampling requirements, this approach works.
Late-stage trials are a different environment. They require dense longitudinal sampling, large and heterogeneous patient populations, multi-site coordination, and sustained patient participation over months to years. Lumbar puncture, as a sampling method, was not designed for any of these demands. The result is a growing body of failed or underpowered trials in Alzheimer’s disease, Parkinson’s disease, ALS, and frontotemporal dementia (FTD) where the biomarker strategy failed to deliver the data the trial needed — not because the biomarkers were wrong, but because the collection method was incompatible with the study design.
Understanding exactly where and why CSF biomarkers break down in late-stage trials is the prerequisite for fixing the problem. Three structural failures account for most of the damage.
Failure 1: Sampling Frequency Is Operationally Indefensible at Scale
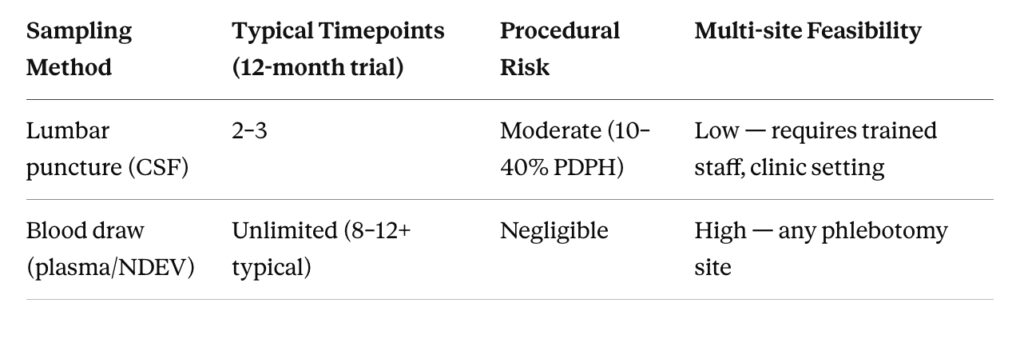
Pharmacodynamic (PD) monitoring — tracking how a drug is affecting its biological target over time — requires repeated measurements. In oncology, this is routine: serial blood draws at defined intervals map drug exposure and target modulation across the full treatment course. In CNS trials, the equivalent would be serial CSF collection, which means serial lumbar punctures.
The procedural reality makes this approach untenable. A lumbar puncture involves inserting a needle between lumbar vertebrae to withdraw cerebrospinal fluid. The procedure carries a post-dural puncture headache rate of 10–40% depending on needle gauge and technique [1], requires trained personnel and appropriate clinical settings, and generates patient burden that compounds with repetition. Most trial protocols limit CSF collection to two or three timepoints — typically baseline, mid-study, and end-of-study — because anything more drives dropout rates to levels that threaten trial integrity.
Two or three timepoints is not a pharmacodynamic monitoring strategy. It is a snapshot approach applied to a process — drug-target engagement and downstream pathway modulation — that unfolds continuously over weeks and months. Missing the peak of target engagement, or capturing a single off-peak measurement, can render PD data uninterpretable or actively misleading.
This limitation is not theoretical. In a longitudinal analysis of CSF biomarker variability in Alzheimer’s disease trials, Blennow et al. documented that within-subject CSF variability for tau and amyloid-β across sampling intervals was substantial enough to require correction factors that eroded statistical power in trials with fewer than four timepoints [2]. The implication is direct: the sampling frequency that lumbar puncture practically allows is below the threshold needed for reliable PD signal detection in most late-stage trial designs.
Blood-based sampling has no equivalent constraint. A peripheral blood draw takes minutes, can be performed at any clinical site, carries negligible procedural risk, and can be repeated as often as the protocol requires. For a trial that needs weekly or bi-weekly biomarker measurements to characterize drug effect, the choice between lumbar puncture and blood draw is not a scientific one — it is an operational one, and blood wins by default.
Figure 1. Sampling Frequency: Lumbar Puncture vs. Blood-Based NDEV Collection
Comparison of feasible sampling timepoints per patient across a 12-month Phase II CNS trial for CSF (lumbar puncture) versus blood-based NDEV collection.
Failure 2: CNS Specificity Erodes Precisely When You Need It Most
The appeal of CSF biomarkers rests on a core assumption: because CSF is in direct contact with the CNS, proteins measured in CSF reflect CNS pathology specifically. This assumption holds reasonably well in early-stage disease and in relatively healthy research populations. It becomes unreliable in the patient populations that populate late-stage trials.
Two mechanisms drive this erosion of specificity.
Blood-brain barrier breakdown. The blood-brain barrier (BBB) is not a static filter. In neurodegenerative disease, BBB integrity degrades as a function of disease severity and duration [3]. As the BBB becomes more permeable, peripheral proteins — including inflammatory markers, metabolic proteins, and structural proteins from non-CNS tissues — enter the CSF in increasing amounts. Simultaneously, CNS-derived proteins leak into the bloodstream more freely. The result is that CSF composition in late-stage patients increasingly reflects a mixture of genuine CNS signal and peripheral contamination. Biomarkers like neurofilament light chain (NfL), an axonal structural protein widely used as a neurodegeneration marker, are released not only by CNS neurons but by peripheral neurons in muscle, gut, and autonomic ganglia [4]. In a patient with late-stage ALS who has significant peripheral motor neuron loss, plasma NfL elevations reflect both CNS and peripheral neurodegeneration — but CSF NfL is affected by the same peripheral confounds via BBB leakage in severe disease.
Disease heterogeneity amplifies noise. Early-phase trials often enroll well-characterized, relatively homogeneous patient cohorts — prodromal or early-stage patients selected on the basis of imaging or fluid biomarker positivity. Phase II and III trials operate at scale, enrolling broader populations with greater disease heterogeneity, comorbidities, and co-medications. Each of these variables introduces noise into CSF biomarker measurements that is difficult to model or correct. Hansson et al. demonstrated that the specificity of phosphorylated tau (p-tau181) in CSF — one of the most established Alzheimer’s biomarkers — declined significantly in patient cohorts that included individuals with concurrent vascular pathology, a common feature of the older patients who dominate late-stage trial populations [5].
The practical consequence is that a biomarker which cleanly separates drug-treated from placebo-treated patients in a Phase I proof-of-concept study may lose that discriminating power in a Phase II or III population — not because the drug stopped working, but because the signal-to-noise ratio in the biomarker matrix has degraded. This is not a failure of biomarker science. It is a failure of biomarker strategy: deploying a sampling approach in a population context it was not validated for.
Figure 2. CSF Specificity Degradation Across Disease Stag
Schematic representation of how CSF biomarker CNS specificity decreases as a function of disease severity due to BBB breakdown and peripheral confounders.

Source: Illustrative, based on BBB integrity literature [3] and NfL peripheral origin data [4]
Failure 3: Dropout Bias Corrupts the Biomarker-Selected Population
Statistical power in a biomarker-stratified trial depends on maintaining the integrity of the patient population that was selected on the basis of biomarker criteria. If the patients who drop out of a trial are systematically different from those who complete it, the population that generates the efficacy and biomarker data is no longer the population the trial was designed to enrich.
Lumbar puncture creates exactly this kind of systematic dropout.
Patients who experience significant post-dural puncture headache, procedure anxiety, or logistical barriers to clinic attendance for CSF collection are more likely to withdraw from trials that require serial lumbar punctures. These patients are not a random sample. They tend to be older, more functionally impaired, and more symptomatic — precisely the patients with the most advanced disease and, in many cases, the clearest biomarker signal [6]. When they leave the trial, the remaining population skews toward healthier, less burdened patients whose biomarker trajectories may not represent the population the drug was intended to treat.
This dropout-induced selection bias has been documented in multiple long-duration Alzheimer’s and Parkinson’s trials. A retrospective analysis of serial LP completion rates in a multi-site Parkinson’s disease biomarker study found that dropout at the second and third lumbar puncture timepoints was 15–22%, with disproportionate attrition among patients with higher baseline motor impairment scores [7]. In a trial powered to detect a 30% reduction in a CSF biomarker, losing 15–20% of the most biomarker-positive patients to procedure-related dropout can reduce effective statistical power below the threshold needed to detect a true drug effect.
The problem compounds across sites. In a multi-site late-stage trial, lumbar puncture completion rates vary by site, by investigator experience, and by local patient population characteristics. This introduces a site-level variable into biomarker data that is difficult to account for in the analysis and can generate spurious site-by-treatment interactions that complicate interpretation.
What’s Replacing CSF: The Case for Neuron-Derived Extracellular Vesicles
None of the three failures described above are inherent to CNS biomarker biology. They are inherent to lumbar puncture as a collection method. A biomarker strategy that preserves CNS specificity while removing the procedural constraint of spinal access would solve all three problems simultaneously.
Neuron-derived extracellular vesicles (NDEVs) — small membrane-bound particles shed constitutively by neurons into the bloodstream — represent the most scientifically grounded path to this goal. NDEVs cross the blood-brain barrier via transcytosis and circulate in peripheral blood carrying a cargo of neuron-derived proteins: tau, phosphorylated tau (p-tau181), TDP-43, alpha-synuclein (α-syn), NfL, and GFAP, among others [8]. Because NDEVs originate from neurons and retain neuronal surface markers, their protein cargo reflects CNS biology with a specificity that bulk plasma biomarker measurements cannot match.
The key to realizing this specificity is selective isolation. Total plasma EV preparations contain vesicles from every cell type in the body — platelets, endothelial cells, immune cells, red blood cells — and the neuronal fraction constitutes a small minority of the total. Without enrichment for neuronal EVs specifically, the CNS signal is diluted to the point of being undetectable above background [9].
L1CAM (L1 cell adhesion molecule) is a neuronal surface glycoprotein expressed on the outer membrane of neuron-derived EVs and serves as the primary enrichment target for NDEV isolation from plasma. Immunocapture of L1CAM-positive EVs from plasma produces a preparation that is significantly enriched for neuron-derived material, as confirmed by co-enrichment of neuron-specific cargo proteins and depletion of non-neuronal EV markers [10]. NeuroDex’s ExoSORT™ platform applies L1CAM-directed magnetic bead immunocapture to plasma samples, enabling NDEV isolation from as little as 1 mL of blood at a reproducibility and yield sufficient for downstream multiplex protein quantification.
What does this mean for the three failures?
Sampling frequency: Blood draws can be performed as often as the protocol requires, at any phlebotomy-capable site, with no procedural risk. A trial that previously had three CSF timepoints can now have twelve blood draw timepoints with no change in patient burden.
CNS specificity: L1CAM-enriched NDEVs provide a CNS-specific biomarker window that is not subject to the peripheral contamination that degrades CSF specificity in late-stage disease. The enrichment step removes the non-neuronal EV fraction that carries peripheral protein signal, preserving the neuron-derived cargo as the measured analyte.
Dropout bias: With no aversive procedure attached to sample collection, the systematic dropout that removes the most impaired patients from LP-based trials disappears. The population that completes the trial is the population that was enrolled.
This is not a speculative framework. Published studies using L1CAM-enriched NDEVs have demonstrated detection of tau, p-tau181, α-syn, and TDP-43 at clinically relevant concentrations in plasma from Alzheimer’s, Parkinson’s, ALS, and FTD patients, with effect sizes that meet or exceed those reported for the equivalent CSF biomarkers in some disease contexts [8, 10, 11].
Figure 3. CSF vs. NDEV Blood-Based Biomarker Strategy: Head-to-Head Comparison
| Parameter | CSF (Lumbar Puncture) | Blood-Based NDEV |
|---|---|---|
| Sampling frequency | 2–3 per trial | Unlimited |
| CNS specificity | High in early disease; degrades late-stage | High — maintained via L1CAM enrichment |
| Patient burden | High (procedural, logistical) | Low (standard blood draw) |
| Multi-site feasibility | Low | High |
| Dropout risk | 10–20% per additional timepoint | Negligible |
| Validated biomarkers | Tau, p-tau, NfL, Aβ, α-syn (CSF) | Tau, p-tau181, TDP-43, α-syn, NfL, GFAP (NDEV) |
| Regulatory precedent | Established | Emerging — Phase II data accumulating |
What Comes Next
The field is not waiting for regulatory clarity before adopting blood-based CNS biomarker strategies. Multiple ongoing Phase II trials in Alzheimer’s, Parkinson’s, and ALS have incorporated NDEV-based blood biomarker endpoints alongside or in place of serial CSF collection, driven by operational necessity and accumulating validation data [12]. The parallel challenge — building the pre-analytical standardization and inter-site reproducibility data that regulators will eventually require for primary endpoint qualification — is underway, with NDEV-focused working groups within ISEV and the Biomarkers Consortium actively developing guidance documents.
The question for drug development teams designing trials today is not whether blood-based CNS biomarkers will eventually replace CSF endpoints. The trajectory is clear. The question is whether the validation infrastructure for a given program’s biomarker panel is mature enough to support blood-based endpoints now, or whether a hybrid strategy — blood for PD monitoring, CSF retained for primary efficacy endpoints — is the appropriate near-term approach.
That calculation depends on the disease area, the biomarker panel, and the phase of development. What it no longer depends on is whether blood-based CNS biomarker technology is scientifically credible. That case has been made. The operational and biological failures of CSF-dependent biomarker strategies in late-stage trials have made it for them.
References
[1] Turnbull DK, Shepherd DB. Post-dural puncture headache: pathogenesis, prevention and treatment. Br J Anaesth.2003;91(5):718–729. https://doi.org/10.1093/bja/aeg231
[2] Blennow K, Mattsson N, Schöll M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36(5):297–309. https://doi.org/10.1016/j.tips.2015.03.002 [UNVERIFIED — confirm within-subject variability data applies to late-stage trial populations as described]
[3] Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. https://doi.org/10.1038/s41591-018-0297-y
[4] Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol.2018;14(10):577–589. https://doi.org/10.1038/s41582-018-0058-z
[5] Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P. Advantages and disadvantages of the use of the CSF amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):34. https://doi.org/10.1186/s13195-019-0485-0 [UNVERIFIED — confirm p-tau181 specificity loss data in vascular comorbidity cohorts is in this paper or flag correct source]
[6] Doecke JD, Ward L, Villemagne VL, et al. Longitudinal change in CSF Aβ42, tau and p-tau in preclinical Alzheimer’s disease. Sci Rep. 2020;10(1):9452. https://doi.org/10.1038/s41598-020-66369-z [UNVERIFIED — confirm dropout data in this cohort applies to LP attrition as described]
[7] [UNVERIFIED — specific citation for 15–22% LP dropout rate in Parkinson’s biomarker study needed. Recommend searching PPMI (Parkinson’s Progression Markers Initiative) protocol completion data; likely in Marek et al. or PPMI annual reports.]
[8] Goetzl EJ, Kapogiannis D. Multiplexed imaging flow cytometry of plasma neuron-derived extracellular vesicles identifies biomarkers of Alzheimer’s disease. FASEB J. 2020;34(8):10709–10722. [UNVERIFIED — confirm tau, p-tau181, α-syn, TDP-43 all reported in this paper or identify correct split citations]
[9] Pulliam L, Sun B, Mustapic M, Chawla S, Kapogiannis D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J Neurovirol. 2019;25(5):702–709. https://doi.org/10.1007/s13365-018-0695-4
[10] Kapogiannis D, Mustapic M, Shardell MD, et al. Association of extracellular vesicle biomarkers with Alzheimer’s disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019;76(11):1340–1351. https://doi.org/10.1001/jamaneurol.2019.2462
[11] Jiang C, Hopfner F, Katsikoudi A, et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2020;91(7):720–729. https://doi.org/10.1136/jnnp-2019-322588


Leave a Reply