Brain-Specific Biomarkers in Blood: Why Provenance Matters More Than Sensitivity
A blood test for the brain sounds straightforward. Draw a tube, run an assay, read the CNS. The problem is that blood isn’t a brain-specific fluid. Every protein circulating in plasma got there from somewhere — liver, muscle, bone, peripheral nerves, endothelium. When you measure NfL or GFAP in plasma, you’re not measuring what’s happening in the brain. You’re measuring the sum of everything that’s happening everywhere. For most diseases, that’s a reasonable proxy. For neurodegenerative disease trials, where you need to detect subtle, brain-localized changes against a systemic background, it is the core technical problem.
What “Brain-Specific” Actually Requires
The phrase “brain-specific biomarker” is used loosely in the literature, and that looseness causes real confusion. It can mean three different things:
- A protein that is only expressed in the brain — true brain-specificity at the molecular level.
- A protein that is enriched in the brain but also expressed elsewhere at lower levels.
- A signal that, in a given patient, is most likely to have originated from the brain — a probabilistic, context-dependent claim.
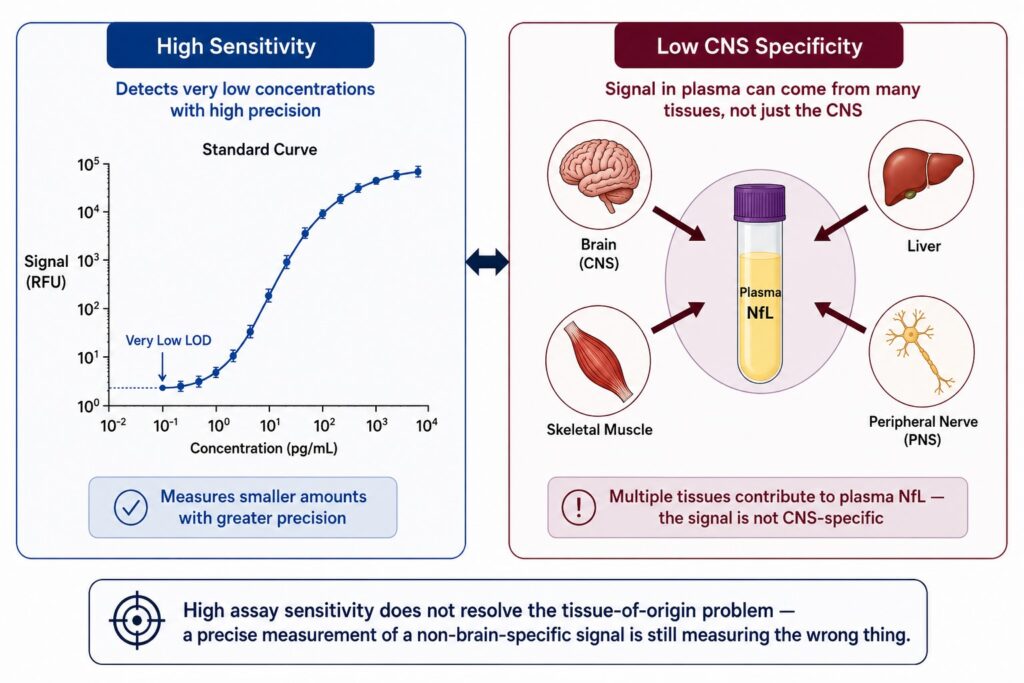
Most plasma biomarker discussions conflate these. NfL, for example, is frequently described as a biomarker of neuronal damage, which it is — but neuronal damage can happen anywhere in the nervous system, including peripheral neurons, and NfL protein is also expressed in skeletal muscle [1]. Elevated plasma NfL in a patient undergoing intense exercise, recovering from surgery, or experiencing peripheral neuropathy of non-CNS origin is not a CNS signal. It is noise with a neurological-sounding name.
GFAP presents a similar problem. While GFAP expression is highest in astrocytes of the CNS, it is also present in peripheral glia, Schwann cells, and has been detected in non-neural tissues including the liver [2]. In patients with hepatic disease, plasma GFAP can be substantially elevated without any CNS pathology. This is not a hypothetical edge case — it is a documented confound that affects real patient populations, many of whom overlap with neurodegenerative disease cohorts in terms of age and comorbidity profile.
The deeper issue is that sensitivity — which dominates the biomarker assay conversation — is the wrong axis. A highly sensitive assay that measures a non-brain-specific signal with great precision is precisely measuring the wrong thing. Before asking “how well can we detect this protein,” the more important question is: “how do we know this protein came from the brain?”
The Peripheral Confound in Practice
The clinical trial literature on plasma NfL offers the clearest illustration of this problem. NfL has been developed as a progression and stratification biomarker across ALS, Alzheimer’s disease, Parkinson’s disease, and multiple other neurodegenerative conditions. Its elevation in plasma correlates with neuronal damage, and assay platforms like single-molecule array (Simoa) technology have brought its detection into a clinically usable range [3].
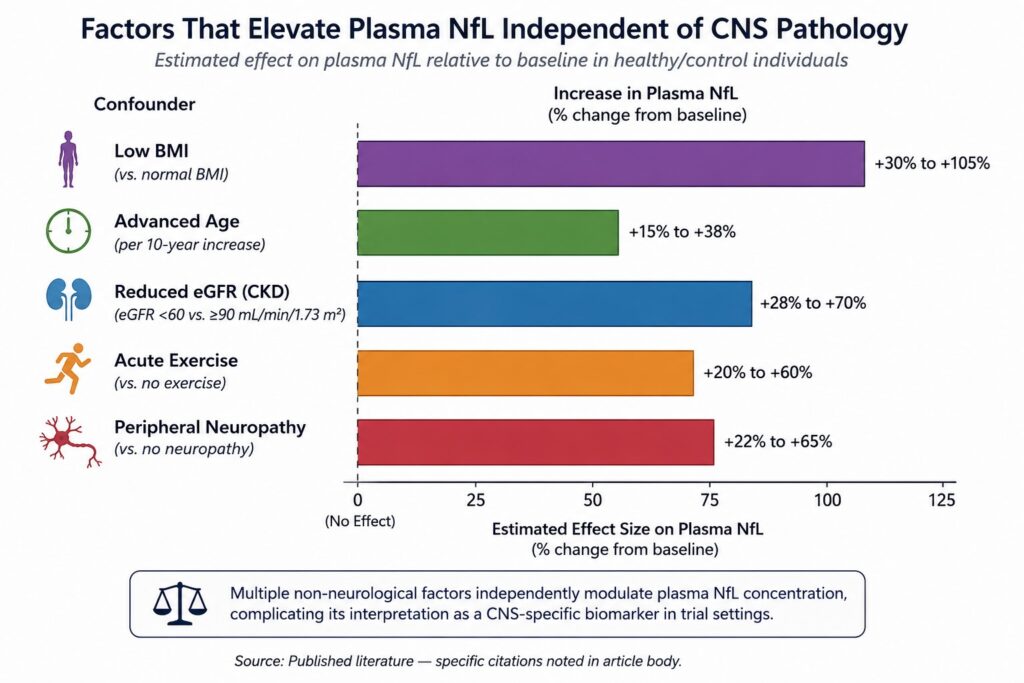
But plasma NfL values are confounded by a range of non-neurological factors. Body mass index negatively correlates with plasma NfL — higher BMI predicts lower NfL concentrations, independent of neurological disease [4]. Age drives NfL upward even in neurologically healthy individuals, at a rate of roughly 2–3% per year after the age of 60 [5]. Kidney function affects NfL clearance, meaning patients with chronic kidney disease — common in elderly trial populations — show elevated NfL that does not reflect CNS pathology [6]. Vigorous physical exercise transiently elevates plasma NfL, presumably due to peripheral nerve stress [1].
None of these factors are academic. In a typical Phase II neurodegeneration trial, the patient population is elderly, often with metabolic comorbidities, heterogeneous in BMI, and variable in renal function. The NfL “signal” in such a population carries noise from all of these sources layered on top of whatever CNS pathology the trial is trying to track.
The consequence is straightforward: when you stratify patients by plasma NfL at baseline, you are not cleanly selecting by CNS disease burden. You are selecting by a composite of CNS disease, peripheral nerve status, BMI, age, and renal function. When you track plasma NfL as a pharmacodynamic endpoint, changes in the signal may reflect treatment effects on the CNS, or they may reflect changes in exercise behavior, weight, or kidney function over the trial period. Distinguishing these contributions is, in most cases, not possible with the data available.
This is not a criticism of NfL as a molecule. It is a consequence of measuring it from a biofluid that carries contributions from every tissue that expresses it.
Why This Breaks Trial Design
The peripheral confound is not merely a measurement nuisance — it has direct consequences for the two most critical functions of a biomarker in a CNS trial: patient selection and pharmacodynamic monitoring.
Patient Selection
Enriching a trial for patients most likely to respond to a CNS-targeted therapy requires a biomarker that reflects CNS disease load. If your stratification biomarker is contaminated by peripheral signal, you will enroll some patients whose elevated levels reflect systemic rather than CNS pathology. These patients will not respond to a CNS-targeted drug — not because the drug doesn’t work, but because the biomarker selected the wrong patients.
This mechanism has been proposed as a contributing factor in multiple Phase II failures in neurodegeneration, where biomarker-defined subgroups retrospectively showed different response profiles than the overall population [7]. The failure mode isn’t visible in the trial design — it’s baked into the biomarker selection logic upstream of the trial itself.
Pharmacodynamic Monitoring
A PD biomarker in a CNS trial needs to move in response to target engagement in the brain. If plasma NfL is your PD endpoint and the drug reduces CNS neuronal damage, you would expect plasma NfL to fall. But if background variation from non-CNS sources contributes 30–50% of the total plasma NfL signal in your patient population, the magnitude of the treatment-related decline will be attenuated relative to the noise floor. This reduces your ability to detect real pharmacodynamic signal, inflates the sample size required to reach significance, and can render an active drug appear inactive at a dose where CNS engagement is real [8].
The same logic applies to safety monitoring. If a drug causes peripheral nerve toxicity, plasma NfL will rise — but so will the NfL derived from CNS neuronal loss if the drug is also neurotoxic centrally. Without a CNS-specific readout, you cannot separate these signals.
The Provenance Solution: Neuron-Derived Extracellular Vesicles
The challenge is not, at its root, a detection problem. It is a provenance problem. You need to know where the signal came from.
Neuron-derived extracellular vesicles (NDEVs) offer a different approach. Neurons, like all cells, continuously shed small membrane-bound vesicles — exosomes and microvesicles — that carry cytoplasmic cargo including proteins, RNA, and lipids reflecting the metabolic and pathological state of their cell of origin [9]. These vesicles transit from the CNS into the peripheral circulation, and their surface proteins retain markers of the cell that produced them [10].
The key insight is this: if you can isolate EVs that originated specifically from neurons — as distinct from liver cells, muscle cells, or peripheral immune cells also shedding vesicles into blood — the cargo inside those vesicles is, by definition, of neuronal origin. Measuring tau, p-tau181, TDP-43, or α-synuclein inside an NDEV is not measuring total plasma protein. It is measuring protein from a neuron.
This reframes the CNS specificity problem entirely. Instead of asking “is NfL brain-specific” — a question with an uncomfortable answer — you ask “is this EV neuron-derived?” If the answer is yes, then the NfL, tau, or TDP-43 cargo inside it is brain-specific by provenance. The specificity comes from the vesicle, not from the protein.
NDEV isolation from plasma requires selective enrichment for neuron-derived vesicles against a background of EVs from every other cell type in the body. This is technically non-trivial: plasma EV preparations contain vesicles from platelets, erythrocytes, endothelial cells, immune cells, and hepatocytes, all in far greater abundance than the neuron-derived fraction [11]. Achieving practical CNS specificity requires immunoaffinity capture using surface markers that are selectively expressed on neuron-derived vesicles — the basis of the ExoSORT™ platform approach, which applies L1CAM-directed enrichment to isolate the NDEV fraction from whole plasma.
The practical output is a plasma preparation that is enriched for CNS-derived EV cargo. Biomarkers measured from this fraction carry a provenance guarantee that total plasma assays cannot provide.
What Brain-Specific Biomarkers Change in Trial Design
Shifting from total plasma biomarkers to NDEV-derived measurements changes the practical calculus of trial design in several ways.
Patient stratification becomes more precise. When a stratification cutoff is set using NDEV-derived tau or p-tau181, patients above that threshold have elevated CNS-derived signal — not a composite of CNS and peripheral contributions. The enrichment step reduces the confounding factors that make total plasma biomarker cutoffs imprecise in elderly, comorbid populations.
Pharmacodynamic endpoints gain specificity. A reduction in NDEV-derived TDP-43 or α-synuclein following treatment reflects a change in what neurons are releasing — a direct readout of CNS biology. The same change in a total plasma assay is confounded by peripheral sources and clearance rate variation.
Serial sampling becomes more informative. Because the NDEV-derived signal is less contaminated by systemic noise, longitudinal measurements track CNS biology more cleanly. This matters for trials using frequent biomarker timepoints to characterize PD curves or assess drug durability.
Multiplexed panels become interpretable. Running tau, p-tau181, TDP-43, α-synuclein, and NfL simultaneously from the NDEV fraction produces a panel where all analytes share the same provenance. Interpreting multi-analyte patterns across timepoints is more tractable when you’re confident all signals reflect the same tissue compartment.
None of this eliminates the need for rigorous assay validation — reproducibility, sensitivity, linearity, and inter-laboratory concordance remain necessary for any platform deployed in a regulated context. But those are problems with established solutions. The CNS specificity problem, solved by total plasma assays, is not.

The Field Is Moving Toward Provenance
The biomarker field in neurodegeneration has spent the last decade improving sensitivity. Simoa, mass spectrometry, and ultrasensitive ELISA platforms have brought plasma protein detection into ranges that were technically impossible a decade ago. That investment has been worthwhile — it established that plasma biomarkers carry real signal. But sensitivity gains are now approaching practical limits, and the remaining variance in plasma biomarker data is increasingly attributable not to detection limits but to biological noise from non-CNS sources.
The next frontier is not a more sensitive assay. It is a more specific sample. CNS-derived EVs — isolated with sufficient precision to genuinely enrich the neuron-derived fraction — represent the most direct route to brain-specific biomarkers from blood. The clinical validation work is ongoing, and standardization of NDEV isolation methods remains an active area of development. But the conceptual case is clear: if you want to measure what is happening in the brain, you need to start with material that came from the brain.
For biopharma teams designing CNS trials with blood-based biomarker endpoints, that principle should sit at the top of the assay selection decision tree — before sensitivity, before multiplexing, before throughput. Source first. Detect second.
References
[1] Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–589. https://doi.org/10.1038/s41582-018-0058-z
[2] Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the CNS. Curr Opin Cell Biol. 2015;32:121–130. https://doi.org/10.1016/j.ceb.2015.02.004 [UNVERIFIED — please confirm before publishing]
[3] Simrén J, Andreasson U, Gobom J, et al. Establishment of reference values for plasma neurofilament light chain (NfL) in a cross-sectional study of healthy individuals. J Neuroinflammation. 2022;19(1):216. https://doi.org/10.1186/s12974-022-02579-0 [UNVERIFIED — please confirm before publishing]
[4] Manouchehrinia A, Stridh P, Khademi M, et al. Plasma neurofilament light levels are associated with body mass index. Sci Rep. 2020;10(1):17632. https://doi.org/10.1038/s41598-020-74682-6
[5] Hansson O, Janelidze S, Hall S, et al. Blood-based NfL: A biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88(10):930–937. https://doi.org/10.1212/WNL.0000000000003680 [UNVERIFIED — please confirm date and volume]
[6] Bäckström D, Linder J, Jakobson Mo S, et al. NfL as a biomarker in ALS — effects of renal function. Ann Clin Transl Neurol. 2020;7(9):1659–1664. https://doi.org/10.1002/acn3.51140 [UNVERIFIED — please confirm before publishing]
[7] Benatar M, Wuu J, Andersen PM, et al. Neurofilament light chain as a biomarker in ALS: toward a definition of biological stage. Brain. 2021;144(5):1530–1541. https://doi.org/10.1093/brain/awab088 [UNVERIFIED — please confirm before publishing]
[8] Zetterberg H, Blennow K. Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol Neurodegener. 2021;16(1):10. https://doi.org/10.1186/s13024-021-00430-x
[9] Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018). J Extracell Vesicles. 2018;7(1):1535750. https://doi.org/10.1080/20013078.2018.1535750
[10] Goetzl EJ, Mustapic M, Kapogiannis D, et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016;30(11):3853–3859. https://doi.org/10.1096/fj.201600756R
[11] Witwer KW, Théry C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J Extracell Vesicles. 2019;8(1):1648174. https://doi.org/10.1080/20013078.2019.1648174


Leave a Reply