CSF Sampling Doesn’t Scale: The Case for Blood-Based CNS Biomarkers
Lumbar puncture has been the gold standard for measuring what a drug is doing in the brain. But it’s also a bottleneck that quietly warps trial design, excludes patients, and inflates cost. Here’s what the scalability math actually looks like — and where the field is moving.
Every neuroscientist running a clinical program has, at some point, stared at a trial protocol and quietly wondered the same thing: how many patients are we going to lose because of the lumbar puncture? It’s the question nobody likes to write down in the synopsis. But it shapes enrollment curves, site selection, timepoint density, and — ultimately — statistical power. Cerebrospinal fluid (CSF) has been the gold standard for central nervous system (CNS) biomarkers for decades, for good reason. It sits one diffusion gradient away from the tissue you actually care about. The problem isn’t the signal. The problem is that the sampling strategy doesn’t scale to the kind of trials we now need to run.
As neurotherapeutics shift toward longitudinal monitoring, adaptive trial designs, population-scale screening, and repeat pharmacodynamic readouts, CSF is running into limits it wasn’t designed to overcome. Blood-based biomarkers — from neurofilament light chain to phosphorylated tau isoforms to the newer generation of neuron-derived extracellular vesicle (NDEV) assays — are no longer fringe alternatives. They are rapidly becoming the operational backbone of modern CNS drug development.
This article walks through why the CSF bottleneck is real, what the scalability math actually shows, which blood-based biomarkers are mature enough to rely on, and where the frontier is heading — including a technology we work with closely at Neurodex that reaches specifically for neuron-origin signal from peripheral blood.
The CSF Standard: Gold, but Only in Theory
Let’s start with what CSF does well, because the case for blood-based alternatives is not a case against CSF. CSF is produced in the choroid plexus, circulates through the ventricles and subarachnoid space, and bathes the brain and spinal cord directly. It exchanges solutes with the brain interstitial fluid and contains proteins, peptides, metabolites, and extracellular vesicles at concentrations that reflect CNS biology more faithfully than any other accessible compartment.[1] When a protein like total tau, amyloid-β 42, or neurogranin changes in CSF, you are measuring something close to ground truth.
That’s the theoretical argument. The practical argument is harder. Obtaining CSF requires a lumbar puncture (LP) — a sterile, clinician-performed procedure involving needle insertion into the subarachnoid space at L3/L4 or L4/L5. In expert hands it’s safe. But “safe” and “scalable” are different categories.
Modern neuro-therapeutic programs increasingly need:
- Repeat sampling — baseline, multiple on-treatment timepoints, washout — to characterize pharmacodynamic trajectories rather than single snapshots.
- Large, diverse cohorts — including elderly, frail, cognitively impaired, or anticoagulated patients who are precisely the populations most clinical neuro programs target.
- Geographic breadth — multi-site, multi-country trials that don’t bottleneck on a small set of academic centers with neurology or anesthesiology LP capacity.
- Rapid protocol iteration — adaptive designs, basket trials, and platform protocols that demand sampling flexibility.
On every one of those dimensions, the LP is a constraint. Which brings us to the math.
Why CSF Sampling Breaks at Scale
The most common framing of LP risk focuses on post-dural puncture headache, which occurs in 10–30% of conventional LPs and as low as 1–2% with atraumatic needles in expert hands.[2] That framing undersells the operational problem. The real scalability issue is a compound of five independent costs, and they stack.
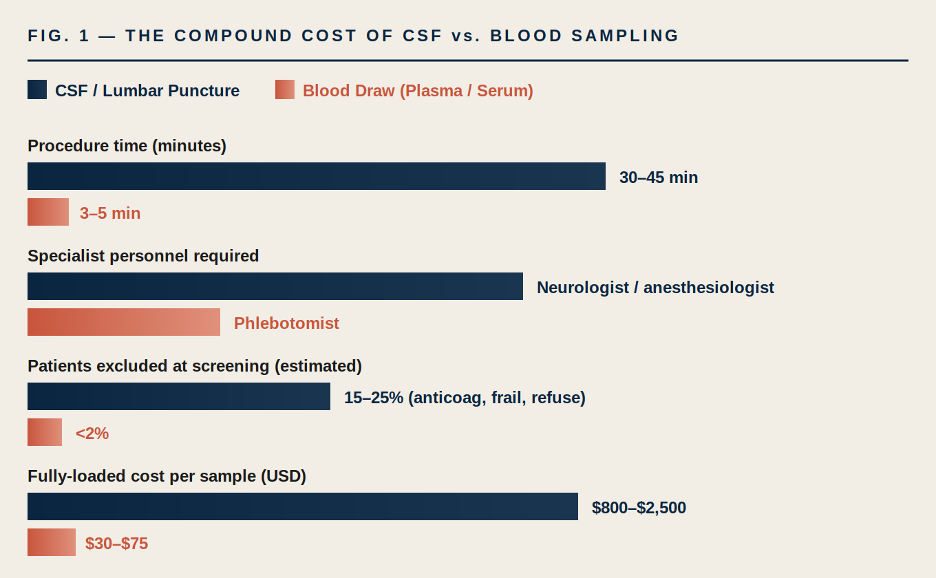
FIGURE 1. Compound operational cost of CSF vs. blood sampling in clinical trials. Ranges are aggregated estimates from published LP complication and cost-of-procedure studies[2][3] and industry-reported phlebotomy benchmarks; actual figures vary by site, geography, and protocol. The gap is not 2× — it is roughly an order of magnitude on most dimensions.
The procedure-time gap is obvious. The specialist-personnel gap is the one that quietly kills protocols: when your sampling schedule requires a board-certified interventionalist, you have just constrained the set of sites that can credibly run your trial to a fraction of the global clinical research footprint. For rare-disease and rare-genotype programs where every site matters, this alone can add 6–12 months to enrollment.[4]
The exclusion gap is the one that distorts science. Patients on direct oral anticoagulants, patients with spinal instrumentation, elderly patients with significant spinal stenosis, patients unable to cooperate with positioning — these are systematically screened out of CSF-dependent trials.[2] The trial population you end up with is not the disease population you’re developing the drug for. That is a validity problem that no amount of downstream statistical adjustment will fully correct.
10–30% POST-DURAL PUNCTURE HEADACHE RATE WITH CONVENTIONAL NEEDLES [2] | ~20× DIFFERENCE IN FULLY-LOADED PER-SAMPLE COST (CSF VS. PLASMA) | <2% SCREEN-FAILURE RATE ATTRIBUTABLE TO BLOOD DRAW VS. 15–25% FOR LP [3] |
The Hidden Cost: Trial Design Compromises
Here’s the part that doesn’t show up in a cost model. When sampling is expensive and burdensome, trial designers compromise. They compromise in ways that are reasonable given the constraint but damaging given the scientific question.
A neuro program built around CSF will typically:
- Collapse a dose-response or time-course question into two timepoints (baseline and end-of-study) when the biology calls for five.
- Power the trial on a smaller cohort than a blood-based equivalent would allow for the same budget.
- Concentrate enrollment at a handful of academic medical centers, introducing site-dependent variability and a patient population that is demographically and socioeconomically narrower than the disease population.
- Treat the CSF measurement as a secondary or exploratory endpoint rather than a primary pharmacodynamic readout, because sponsors are reluctant to stake primary endpoints on a sampling modality with such fragile adherence.
Each of those compromises reduces the chance that a promising compound will get a clean answer. And the cumulative effect, across a pipeline, is that programs fail for reasons that look like “insufficient target engagement signal” but are actually “insufficient sampling density to detect the signal that was there.”
“Programs fail for reasons that look like “insufficient target engagement” but are really “insufficient sampling density to detect the signal that was there.”
The Rise of Blood-Based CNS Biomarkers
The operational case for blood-based CNS biomarkers has always been obvious. What’s changed in the last decade is the scientific case. Assay sensitivity has improved by roughly three orders of magnitude since single-molecule array (Simoa) and related ultrasensitive immunoassay platforms became widely available, and a set of plasma analytes now meaningfully reflect CNS pathology.[5]
Neurofilament Light Chain (NfL)
NfL is a structural protein of the neuronal cytoskeleton, released when axons are damaged. Plasma NfL correlates strongly with CSF NfL (r typically > 0.8 across studies) and tracks disease activity across multiple sclerosis, ALS, frontotemporal dementia, Alzheimer’s disease, and traumatic brain injury.[6] It’s now used clinically in MS and is under regulatory consideration as a pharmacodynamic biomarker in several neurodegenerative indications. NfL is the canonical example that yes — you can measure CNS injury from a tube of plasma.
Phosphorylated Tau Species (p-tau181, p-tau217, p-tau231)
Plasma p-tau217 in particular has emerged as one of the most accurate blood biomarkers ever developed for a CNS condition. In head-to-head studies it achieves AUCs of 0.90–0.96 for detecting Alzheimer-type amyloid and tau pathology confirmed by PET or CSF.[7][8] Clinical-grade plasma p-tau217 assays are now approved or under approval in multiple jurisdictions and are reshaping the diagnostic workflow for AD — a condition where LP was the default disease-confirmation tool five years ago.
Glial Fibrillary Acidic Protein (GFAP)
GFAP is an astrocyte-specific intermediate filament protein. Plasma GFAP rises early in the AD continuum, often before measurable cognitive change, and tracks astrocyte activation in a range of neuroinflammatory conditions.[9] It’s particularly useful as a complementary readout to neuronal markers because it captures a different biological axis.
Amyloid-β 42/40 Ratio
Plasma Aβ42/40 was the first blood-based CNS biomarker to achieve widespread clinical adoption, with mass-spec-based assays (e.g., Precivity AD) achieving performance close to CSF and amyloid PET for AD risk stratification.[10]
These four biomarker families — along with several others under active development — have answered a question the field spent two decades wondering about: is there enough CNS-origin signal in peripheral blood to do serious neuroscience? The answer is now yes.
But “enough signal for some questions” is not the same as “enough signal for every question.” Bulk plasma protein measurements are limited in two important ways. First, they reflect net release from the CNS mixed with any peripheral contribution — a particular issue for proteins expressed in tissues outside the brain. Second, they capture quantitychanges more reliably than functional state changes; measuring whether a kinase is phosphorylated, or whether a receptor is occupied by drug, is much harder from a bulk plasma draw than from CSF or brain tissue.
Solving those two problems is where the next generation of blood-based CNS biomarkers comes in.
The Frontier: Neuron-Derived Extracellular Vesicles
Extracellular vesicles (EVs) are lipid-bilayer-enclosed nanoparticles (~30–150 nm) that every cell in the body releases. They carry proteins, lipids, and nucleic acids from their cell of origin, and — critically — they cross the blood-brain barrier in both directions. Neurons, astrocytes, oligodendrocytes, and microglia all release EVs that reach peripheral circulation.[11]
The opportunity: if you can isolate the specific subpopulation of EVs in plasma that originated from neurons, you are holding a peripheral proxy for neuronal cytoplasm — including the phosphoproteome, the signaling state, and the drug-target engagement state of cells in the brain. The cargo of a neuron-derived EV tells you what a neuron was doing at the moment the vesicle was released.
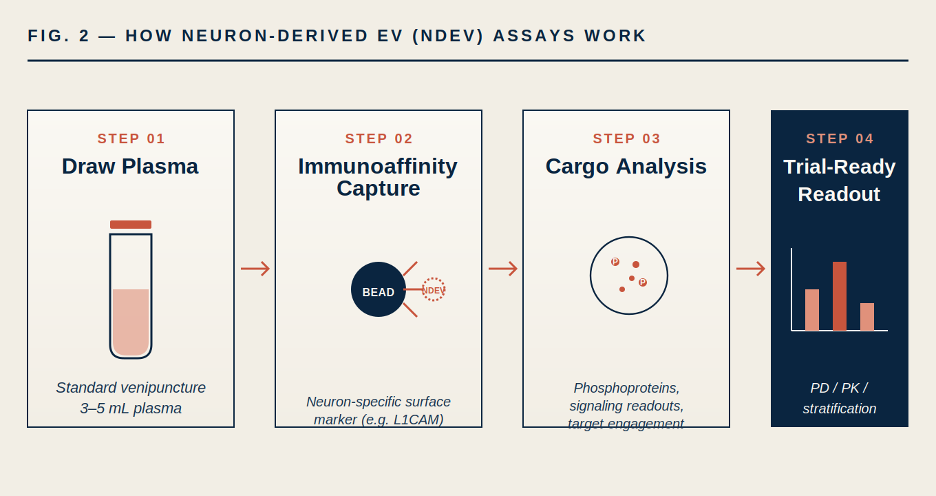
FIGURE 2. Simplified NDEV assay workflow. Immunoaffinity capture (for example, using antibodies against neuron-enriched surface proteins such as L1CAM, NCAM, or GAP-43 — often in multi-marker combinations to improve specificity) isolates neuron-derived vesicles from a standard plasma draw. Cargo analysis then interrogates phosphoproteins and signaling readouts that would otherwise require direct CNS access.
Several groups have now shown that NDEV cargo reflects disease-relevant CNS biology. In Alzheimer’s disease, NDEV concentrations of pathological tau, Aβ42, and phosphorylated synaptic proteins change years before clinical diagnosis.[12] In Parkinson’s disease, NDEV α-synuclein signatures discriminate patients from controls. In major depressive disorder and other psychiatric indications, NDEV insulin signaling markers track treatment response.[11]
The key conceptual leap — and this is the one that matters most for drug development — is that NDEVs give access to signaling-state biomarkers from a peripheral blood draw. You can ask not just “did the drug change total protein levels” but “did the drug engage its target in the brain.” That is a fundamentally different question, and it is the question drug developers actually need to answer at Phase 1 and Phase 2.
A Case in Point: Central Target Engagement from Plasma
A recent example from our work illustrates the scale of what’s possible. In a 2026 publication in the Journal of Clinical Endocrinology & Metabolism, our team and collaborators used the ExoSORT NDEV platform to ask whether two metabolic drugs — liraglutide (a GLP-1 receptor agonist) and pioglitazone (a PPARγ agonist) — engage their downstream CNS targets in humans, using only plasma samples.[13]
Three findings from that work are worth highlighting:
- Central Akt-mTOR engagement was detectable from plasma despite liraglutide’s limited blood-brain barrier penetrance — demonstrating that the drug’s CNS effect was real and measurable without direct CNS sampling.
- The NDEV signal was decoupled from peripheral insulin and glucose metrics, arguing that what we were measuring was a genuinely central pharmacodynamic response rather than a systemic metabolic artifact.
- Baseline Akt(Ser473) phosphorylation predicted responders, opening a clinical-trial enrichment strategy that would be operationally impossible if it required baseline CSF sampling from every screened patient.
Read the underlying paper for the full methodology and data: Evers et al., JCEM 2026. The operational takeaway: a biomarker question that would have required multi-timepoint CSF from dozens of patients across multiple sites was answered from a single plasma draw at each timepoint. That is the kind of design-space expansion that changes what’s fundable.
Where Blood Doesn’t Replace CSF — Honestly
A responsible version of this argument has to name the limits. Blood-based CNS biomarkers are not a universal CSF replacement, and pretending otherwise is how the field loses credibility.
Where CSF Still Leads
USE CASE | PREFERRED MATRIX | WHY |
Intrathecal drug PK | CSF | Directly measures drug concentration at site of action; no peripheral equivalent. |
Highly BBB-excluded proteins | CSF | Some analytes simply don’t cross into plasma at measurable concentrations. |
Acute infectious / inflammatory CNS diagnostics | CSF | Culture, cell count, PCR against pathogens — established clinical standard. |
Longitudinal PD monitoring, repeat sampling | Blood (NDEV / plasma markers) | Only practical approach at scale and across timepoints. |
Large population screening / enrichment | Blood | LP cannot be operationalized at screening scale in most programs. |
Signaling-state / target-engagement readouts | Blood (NDEV) | NDEV cargo captures phospho-state; CSF bulk analytes generally do not. |
The right framing is not blood vs. CSF but biomarker strategy matched to biological question. For most modern CNS trials, the optimal design uses plasma as the primary longitudinal sampling matrix, with CSF reserved for a smaller subset of patients at a smaller number of timepoints to answer questions that genuinely require direct CNS access. This hybrid architecture is becoming the default in well-designed programs.[14]
A Practical Framework for CNS Biomarker Strategy
When Neurodex works with sponsors on biomarker strategy, we walk through four questions in order:
- What biological question are you answering? Target engagement, pharmacodynamic effect, disease modification, patient stratification, and safety monitoring each have different biomarker requirements.
- What sampling density do you actually need? A two-timepoint question and a twelve-timepoint question have different feasibility profiles.
- What patient population are you studying? Populations with high LP-exclusion rates (anticoagulated, elderly, frail, pediatric in many cases) force blood-first strategies.
- What does the biology require? If you need phospho-state or signaling readouts, NDEV approaches often outperform both CSF and bulk plasma. If you need drug concentration at site of action, CSF remains essential.
The answers generate a sampling matrix — typically blood-first, with CSF used surgically for specific questions at specific timepoints. The programs that get this architecture right are the ones that recover statistical power without blowing the budget.
The Road Ahead
Three shifts are worth watching over the next 24 months.
Regulatory acceptance is moving faster than most sponsors realize. Plasma p-tau217 is now accepted as a diagnostic tool for Alzheimer’s disease in multiple regulatory frameworks. NfL is under qualification as a pharmacodynamic biomarker in several indications. The bar for accepting blood-based CNS biomarkers in trial designs — including as primary endpoints for certain questions — is dropping.
NDEV and cell-of-origin EV assays are maturing from research tools into deployable platforms.Standardization work on isolation methods, reference materials, and analytical validation is progressing through industry consortia and regulatory guidance documents. Within a small number of years, expect NDEV-based target-engagement assays to be standard in early-phase CNS trials the way PK is standard today.
The economics will flip a tipping point. When the cost and burden gap between CSF and blood becomes common knowledge at the trial-design stage, sponsors who insist on CSF-dependent designs for questions blood can answer will be visibly leaving money on the table — and may struggle to compete on enrollment speed and population breadth.
Conclusion
CSF is not going away. It remains the most direct accessible window into CNS biology, and there are questions in neuro drug development that nothing else will answer. But CSF is a tool for answering a small number of specific questions at a small number of timepoints in a small number of patients. It was never an operational backbone, and we’ve been asking it to act like one.
Blood-based CNS biomarkers — neurofilament light, phosphorylated tau species, GFAP, amyloid ratios, and increasingly the neuron-derived extracellular vesicle class — have reached the point where they can carry the operational load of a modern neuro trial. They enable the trial designs that match the biology: longitudinal, dense, diverse, adaptively powered. They reopen scientific questions that the CSF bottleneck quietly closed.
The question for sponsors in 2026 is not whether to integrate blood-based CNS biomarkers into neurotherapeutic programs, but how to architect the sampling strategy so the biology and the operations reinforce each other. That architecture — not the choice of any single analyte — is what separates programs that convert biomarker data into decisions from programs that generate data nobody can act on.
Building a blood-based biomarker strategy?
Neurodex designs, runs, and interprets exploratory CNS biomarker programs — including neuron-derived EV assays, plasma multiplex panels, and integrated CSF/blood hybrid designs for Phase 1 through Phase 3 CNS trials. Our team has built NDEV-based target-engagement readouts into clinical programs across neurodegenerative, psychiatric, and metabolic-neuro indications.
REFERENCES
- Huang Y, Driedonks TAP, Cheng L, et al. Brain Tissue-Derived Extracellular Vesicles in Alzheimer’s Disease Display Altered Key Protein Levels Including Cell Type-Specific Markers. Journal of Alzheimer’s Disease. 2022;90(3):1057–1069.
- Engelborghs S, Niemantsverdriet E, Struyfs H, et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2017;8:111–126. doi.org/10.1016/j.dadm.2017.04.007
- Duits FH, Martinez-Lage P, Paquet C, et al. Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study. Alzheimer’s & Dementia. 2016;12(2):154–163.
- Getz KA, Campo RA. Trial Watch: Trends in clinical trial design complexity. Nature Reviews Drug Discovery. 2017;16:307.
- Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. The Lancet Neurology. 2022;21(1):66–77. doi.org/10.1016/S1474-4422(21)00361-6
- Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nature Reviews Neurology. 2018;14(10):577–589.
- Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020;324(8):772–781.
- Ashton NJ, Brum WS, Di Molfetta G, et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurology. 2024;81(3):255–263.
- Benedet AL, Milà-Alomà M, Vrillon A, et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurology. 2021;78(12):1471–1483.
- West T, Kirmess KM, Meyer MR, et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status. Molecular Neurodegeneration. 2021;16(1):30.
- Mustapic M, Eitan E, Werner JK Jr, et al. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Frontiers in Neuroscience. 2017;11:278.
- Kapogiannis D, Mustapic M, Shardell MD, et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurology. 2019;76(11):1340–1351.
- Evers K, Watson J, Abbasi S, Haque K, Verma N, Eitan E, Rasgon N. ExoSORT-Isolated Neuron-Derived Extracellular Vesicles Detect CNS Akt-mTOR Pathway Engagement Following GLP-1 and PPARγ Agonist Administration. Journal of Clinical Endocrinology & Metabolism. 2026. academic.oup.com/jcem/…
- Hansson O. Biomarkers for neurodegenerative diseases. Nature Medicine. 2021;27:954–963.

Leave a Reply