TDP-43 Proteinopathy
The Unifying Driver of Neurodegeneration — and the Race for a Blood Biomarker
How ALS, frontotemporal dementia, Alzheimer’s and LATE converge on a single protein — and why neuron-derived extracellular vesicles may finally make TDP-43 measurable from a blood draw.
Keywords: TDP-43, TDP-43 proteinopathy, ALS biomarker, frontotemporal dementia, LATE, cryptic peptides, seed amplification assay, TDP-43 PET, neuron-derived extracellular vesicles, blood biomarker
For most of the last two decades, amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Alzheimer’s disease (AD) were studied as separate disorders with separate biologies. That consensus is now breaking down. Across neuropathology, biomarker science, and imaging chemistry, one molecule keeps surfacing as the common thread: TDP-43 (TAR DNA-binding protein of 43 kDa), an RNA-binding protein whose mislocalization and aggregation define a proteinopathy that spans a large, underserved patient population.
This article explains what TDP-43 is, why the field increasingly treats its pathology as a single biological entity, why measuring it in biofluids has proven so difficult, and which biomarker strategies — cryptic peptides, seed amplification assays, PET imaging, and extracellular vesicles — are most likely to deliver a clinically usable test.
What is TDP-43, and why does it matter?
TDP-43 is a predominantly nuclear protein that regulates many steps of RNA metabolism, including transcription, splicing, transport, and stability. One of its most important jobs is splicing repression: it suppresses the inclusion of “cryptic” exons — normally silent stretches of sequence — in hundreds of transcripts. In disease, TDP-43 exits the nucleus and accumulates as insoluble cytoplasmic inclusions. The consequence is twofold: a loss of normal nuclear function (cryptic exons are no longer repressed) and a toxic gain of function from aggregated, misfolded protein [1].
The genetic evidence reinforces its central role. Mutations in TARDBP (the gene encoding TDP-43) cause familial ALS, while the two most common genetic causes of ALS-FTD — C9orf72 repeat expansions and GRN (progranulin) mutations — both converge on TDP-43 pathology downstream. TDP-43 also contains a low-complexity domain that drives liquid-liquid phase separation; the same property that lets it form functional condensates is thought to predispose it to the aberrant phase transitions that seed irreversible aggregation. Understanding these mechanisms matters for biomarkers, because each step — mislocalization, loss of splicing repression, aggregation, and secretion — offers a distinct analyte that a test might capture.
One protein across the neurodegeneration spectrum
The reason TDP-43 has become a focal point is simple: its pathology appears far beyond any single diagnosis. Roughly 97% of ALS cases show TDP-43 pathology, and it is the defining lesion of a large fraction of frontotemporal lobar degeneration (FTLD-TDP) [1]. It is also the third most common proteinopathy in Alzheimer’s disease — behind amyloid-β and tau — with TDP-43 co-pathology reported in roughly 40–57% of AD brains, where it associates with worse memory and greater medial-temporal atrophy [4].
Perhaps most striking is LATE — limbic-predominant age-related TDP-43 encephalopathy — defined by an international consensus in 2019 as a distinct, common cause of an amnestic dementia that clinically mimics Alzheimer’s [2]. LATE neuropathological change affects more than 10% of people over 65 and a substantial share of those over 85 [3]. Taken together, TDP-43 pathology touches more than 20 million people worldwide — this is no longer a rare-disease story.
The 2019 LATE consensus also introduced a neuropathological staging system (stages 1–3) tracing the spread of TDP-43 from the amygdala to the hippocampus and then the middle frontal gyrus, often co-occurring with hippocampal sclerosis [2]. Because LATE and Alzheimer’s frequently coexist and produce overlapping amnestic syndromes, they are almost impossible to disentangle at the bedside today. This diagnostic ambiguity is not academic: it means a meaningful fraction of patients enrolled in — or excluded from — Alzheimer’s trials may be driven by a protein those trials never measure.
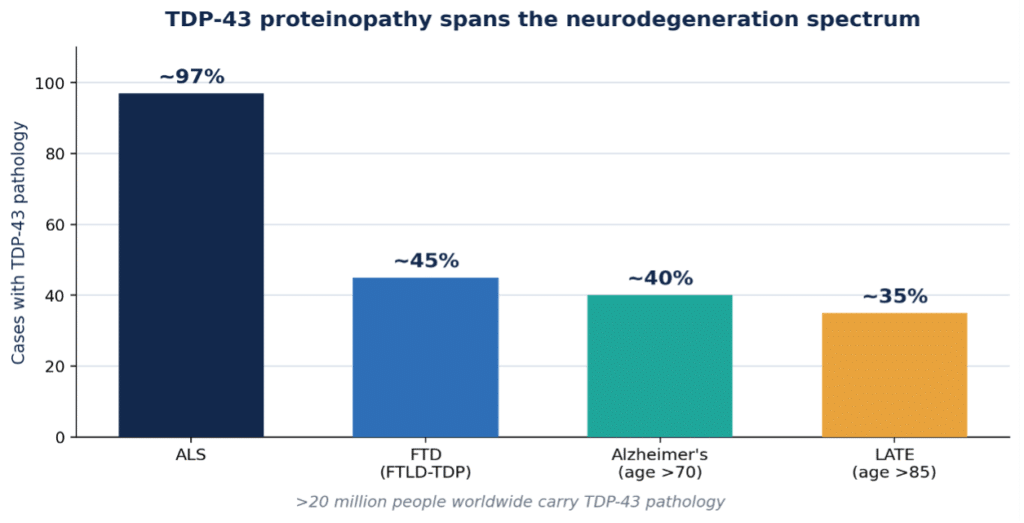
Figure 1. TDP-43 proteinopathy across the neurodegeneration spectrum (original graphic; figures approximate and drawn from refs 1–4).
From symptoms to biology: the case for a unified framework
Fifteen years ago, the Alzheimer’s field shifted from defining disease by symptoms to defining it by underlying biology — amyloid and tau. A parallel move is now being proposed for TDP-43, sometimes framed as TDP-43-Associated Neurodegeneration (TAN): lumping these historically separate diagnoses under one biological definition anchored on the hallmark pathology. The rationale is practical as much as conceptual. A biological definition enables earlier detection, cleaner clinical-trial enrichment, and shared therapeutic targets across ALS, FTD, and TDP-43-driven dementias. But a biological definition is only as good as the tools available to measure the biology — which is where the field hits its central bottleneck.
The Alzheimer’s analogy is instructive. The amyloid/tau framework only became actionable once amyloid PET and, later, blood-based p-tau assays let researchers confirm pathology in living patients, enrich trials with biomarker-positive participants, and read out target engagement. TDP-43 has no equivalent validated tool yet. That gap explains why so much energy at recent scientific meetings has shifted from arguing about nomenclature to solving measurement — a validated TDP-43 biomarker is the rate-limiting reagent for the entire TAN program, from patient stratification to demonstrating that a candidate therapy actually lowers pathology.
The core problem: direct TDP-43 measurement has not delivered
The obvious approach — measure soluble TDP-43 directly in cerebrospinal fluid (CSF) or blood — has repeatedly disappointed. Across assay platforms, soluble TDP-43 in biofluids yields only a weak, inconsistent signal that cannot reliably support differential diagnosis or clinical trials. The pathological protein is largely aggregated and sequestered, the healthy protein is ubiquitous, and blood in particular is dominated by a high-abundance plasma proteome that swamps neuronal signal. As a result, the field has pivoted toward four indirect but more tractable strategies.
There is also a practical dimension to where a biomarker is measured. CSF requires a lumbar puncture, which limits screening, repeat sampling, and use in frail elderly patients — exactly the LATE population. A blood-based test would enable population-scale screening, longitudinal monitoring of progression, and decentralized clinical trials. The prize, therefore, is not merely any TDP-43 signal but one that survives the journey into peripheral blood while remaining specific to neuronal pathology.
Four biomarker strategies gaining ground
1. Cryptic peptides — reading TDP-43’s loss of function
When TDP-43 leaves the nucleus, its splicing-repression function fails and cryptic exons appear in transcripts such as HDGFL2, STMN2, and UNC13A. Some in-frame cryptic exons encode novel peptides that can be detected in biofluids. A landmark Nature Medicine study showed a cryptic HDGFL2 neoepitope is elevated in CSF — and even in presymptomatic C9orf72 carriers — with ultra-high-sensitivity assays reaching limits of detection near 0.016 pg/mL [5], [6]. Because expression varies between patients, groups are building multi-marker panels (HDGFL2, STMN2, UNC13A, and others), and a recent NfL:HDGFL2 ratio outperformed either marker alone for discriminating ALS/FTD from controls and tracked progression [7].
2. Seed amplification assays — catching misfolded conformers
Borrowed from the prion field and already transformative in Parkinson’s and Lewy body dementia, seed amplification assays (SAA / RT-QuIC) use patient-derived misfolded TDP-43 as a template to nucleate aggregation of recombinant monomer, generating an amplified fluorescent readout. New digital SAA formats isolate individual aggregates in nanoliter compartments and have quantified elevated TDP-43 seeds in FTLD-TDP CSF that correlate with severity [8], [9]. The studies remain small and somewhat inconsistent, and a persistent technical hurdle is the reproducible production of recombinant TDP-43 substrate.
3. PET imaging — seeing the pathology in vivo
A TDP-43-selective PET tracer would let clinicians visualize and stage pathology directly. The first-in-class candidate [18F]ACI-19626 was characterized in Nature Communications in 2025, showing selective binding to aggregated TDP-43 over amyloid-β, tau, and α-synuclein; early first-in-human imaging reported higher tracer uptake in expected regions in C9orf72-FTD than in controls [10]. Multiple academic and industry groups are racing to develop competing tracers, though sensitivity to intracellular, conformationally heterogeneous aggregates remains the key challenge.
4. Extracellular vesicles — a route to a blood test
Repeatedly highlighted as one of the most promising paths to a blood biomarker, extracellular vesicles (EVs)concentrate proteins and RNA of neuronal origin and can be immuno-isolated by cell of origin, bypassing the high-abundance plasma background that has defeated conventional assays [11]. Critically, neuron-derived EVs can carry both aggregated TDP-43 and the downstream cryptic-peptide and RNA targets described above — offering multiple orthogonal analytes from a single peripheral draw [14].
The remaining challenges for EV biomarkers are analytical rather than conceptual: reproducible, high-specificity capture of neuron-derived vesicles from plasma, standardized workflows, and demonstrating that the TDP-43 species recovered genuinely reflect CNS pathology rather than peripheral background. These are precisely the problems that determine whether a promising research signal becomes a validated clinical assay — and they are the problems on which the next wave of the field will be judged.
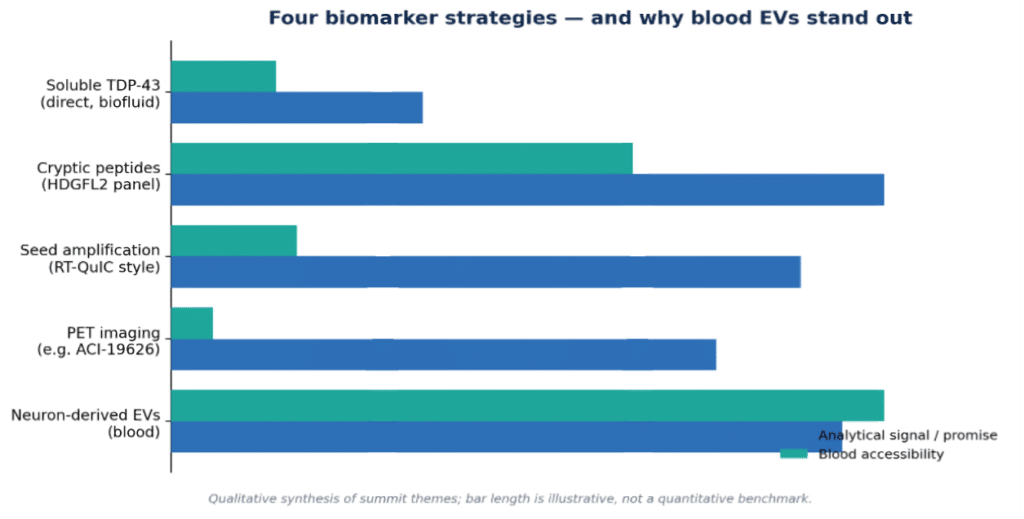
Figure 2. Comparison of biomarker strategies. Bar lengths are a qualitative synthesis, not a quantitative benchmark. See sources: cryptic peptides [5], SAA [8], PET [10], EVs [11].
Where the biology and the biomarker strategy converge
A compelling feature of the EV approach is that it aligns with the underlying cell biology. Work on lysosomal dysfunction shows that TDP-43 fibrils accumulate inside lysosomes, and that secretory autophagy can release that material into the extracellular space — precisely the cargo EVs carry [12]. Dysregulation of the progranulin-driven autophagy–lysosomal pathway likewise drives TDP-43 secretion via EVs [13]. In other words, the same machinery that fails in disease is the machinery that exports TDP-43 into a compartment we can sample from blood. Biology and measurement strategy point in the same direction.
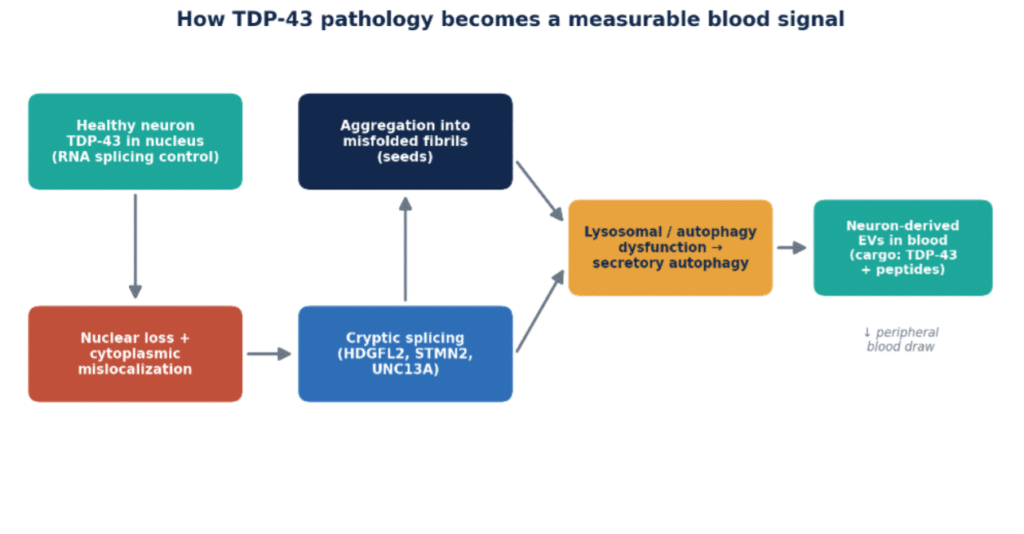
Figure 3. From nuclear TDP-43 loss to a measurable blood signal (original schematic, synthesizing refs 5, 11–13).
Where NeuroDex fits
NeuroDex’s neuron-derived EV isolation platform was built specifically to solve the problem the field keeps returning to: how to measure neuronal pathology from a peripheral blood draw without being defeated by plasma’s high-abundance background. As TDP-43 biology becomes better understood, NeuroDex is positioned to translate each new insight into a blood-based assay.
The platform is already detecting TDP-43 in blood neuron-derived EVs using multiple immunoassays.
The enrichment workflow is a 96-well automated platform with high specificity and reproducibility, and full method validation in a CLIA environment is targeted for the end of 2026.
Contact: info@neurodex.bio · Natick, Massachusetts
References
- Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron / TDP-43 proteinopathy review (PMC). https://pmc.ncbi.nlm.nih.gov/articles/PMC4404432/
- Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503-1527. https://academic.oup.com/brain/article/142/6/1503/5481202
- Limbic-predominant age-related TDP-43 encephalopathy in the oldest old: a population-based study. Brain. 2025;148(1):154. https://academic.oup.com/brain/article/148/1/154/7700928
- Comprehensive assessment of TDP-43 neuropathology data in the National Alzheimer’s Coordinating Center database. Acta Neuropathol. 2024. https://link.springer.com/article/10.1007/s00401-024-02728-8
- Seddighi S, Qi YA, et al. A fluid biomarker reveals loss of TDP-43 splicing repression in presymptomatic ALS-FTD. Nat Med. 2024;30:696-706. https://www.nature.com/articles/s41591-023-02788-5
- Irwin KE, Jasin P, et al. HDGFL2 cryptic proteins report presence of TDP-43 pathology in neurodegenerative diseases. Mol Neurodegener. 2024;19:29. https://link.springer.com/article/10.1186/s13024-024-00718-8
- Light Chain Neurofilament to HDGFL2 cryptic peptide ratio as a fluid biomarker to monitor TDP-43 dysfunction in ALS and FTD. medRxiv. 2025. https://www.medrxiv.org/content/10.64898/2025.12.30.25343222v1
- Borberg E, et al. Digital seed amplification assay for TDP-43 aggregate quantification in CSF. Alzheimers Dement. 2026 (medRxiv 2025). https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/alz.71272
- Ultrasensitive detection of TDP-43 and amyloid-beta protein aggregates using micelle-assisted seed amplification assay (PMC). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11460235/
- Seredenina T, et al. Development of [18F]ACI-19626 as a first-in-class brain PET tracer for imaging TDP-43 pathology. Nat Commun. 2025;16. https://www.nature.com/articles/s41467-025-64540-6
- Extracellular vesicles in TDP-43 proteinopathies: pathogenesis and biomarker potential. Mol Neurodegener. 2025;20. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-025-00859-4
- TDP-43 secretion via extracellular vesicles is regulated by macroautophagy. Autophagy Reports. 2023. https://www.tandfonline.com/doi/full/10.1080/27694127.2023.2291250
- Dysregulation of the progranulin-driven autophagy-lysosomal pathway mediates secretion of the nuclear protein TDP-43 (PMC). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10641265/
- Evaluation of blood-based extracellular vesicles as biomarkers for aging-related TDP-43 pathology (PMC). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9753157/



Leave a Reply