
NfL Has Earned Its Place in CNS Trials, But It’s Not Enough. Here’s What It Can’t Tell You.
Plasma NfL is the most validated blood biomarker in neurodegeneration. Here’s an honest account of what it measures well — and where it stops being useful.
Key Takeaways
- Plasma NfL is a genuine advance in neurodegeneration biomarker science — its track record across ALS, Alzheimer’s, and Parkinson’s research is real and earned.
- NfL has two structural limitations that better assay technology cannot fix: it lacks CNS specificity and it lacks disease specificity.
- NfL rises in any condition causing axonal damage — TBI, MS, chemotherapy neurotoxicity, peripheral neuropathy — making it a marker of neuronal loss, not a marker of a disease.
- In drug trials, NfL cannot stratify patients by disease subtype, distinguish CNS drug effect from peripheral nerve toxicity, or track pathology-specific cargo like TDP-43 or α-synuclein.
- NDEV-based panels that add provenance-controlled, disease-specific analytes alongside NfL address each of these gaps without discarding NfL’s established utility
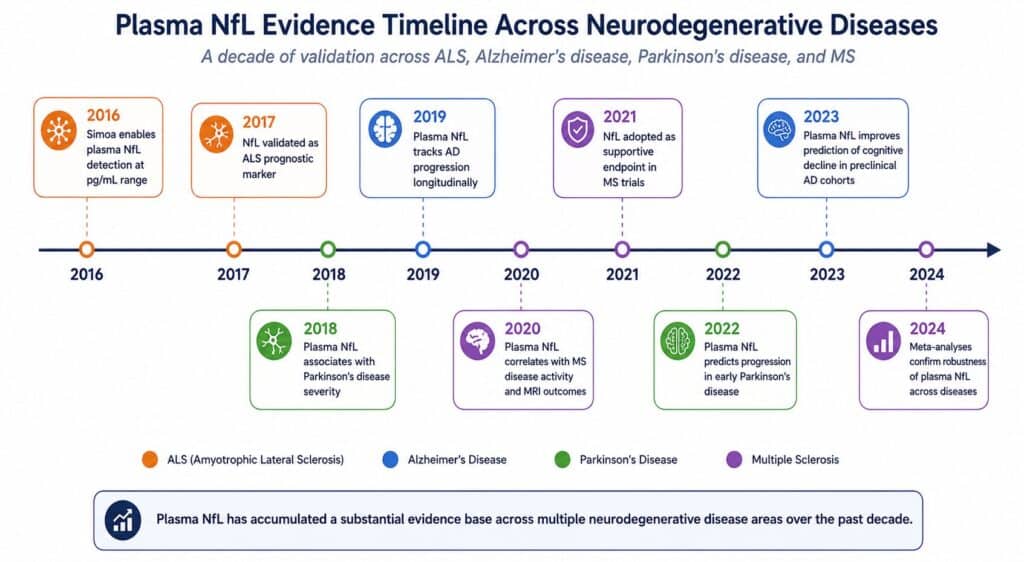
Neurofilament light chain has had a remarkable decade. A cytoskeletal protein largely ignored outside neuropathology labs in the early 2000s, NfL is now one of the most widely measured blood biomarkers in neurodegeneration research, with a literature spanning ALS, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and traumatic brain injury. Ultrasensitive detection platforms brought it into a clinically usable range, longitudinal cohort studies demonstrated its prognostic value, and regulatory bodies have begun accepting it as a supportive endpoint in CNS trials. That trajectory reflects genuine scientific progress.
It also reflects a tendency, common in biomarker science, to extend a useful tool beyond the problems it was built to solve. NfL is excellent at measuring neuronal loss. It is not a measure of where that loss is occurring, what is causing it, or which molecular pathway is driving it. For the questions that now dominate CNS drug development — patient stratification by pathological subtype, pharmacodynamic monitoring of mechanism-specific therapies, distinguishing treatment effect from disease progression — NfL’s structural limitations become the binding constraint.
This is not a critique of NfL. It is an attempt to be precise about what it measures, what it doesn’t, and what needs to sit alongside it in a modern CNS trial biomarker strategy.
What NfL Does Well — And Why the Evidence Is Real
NfL is a subunit of the neurofilament complex, a cytoskeletal scaffold that maintains axonal structure and regulates conduction velocity in neurons [1]. When axons are damaged — through degeneration, inflammation, or mechanical injury — neurofilaments are released into the extracellular space, enter the CSF, and cross into peripheral blood. The plasma concentration of NfL therefore tracks the rate of neuroaxonal damage across the nervous system.
This basic biology has proven clinically useful in several well-documented contexts.
Prognostic stratification in ALS. Plasma NfL concentration at diagnosis correlates with ALS progression rate and survival. Patients with higher baseline NfL decline faster and live shorter [2]. This association is strong enough that NfL has been proposed as an enrichment biomarker for ALS trials — selecting faster progressors for trials of neuroprotective agents where a treatment signal is more detectable against a higher background rate of change.
Longitudinal disease monitoring. In Alzheimer’s disease, plasma NfL rises with disease stage and tracks cognitive decline longitudinally [3]. In multiple sclerosis, NfL drops rapidly in response to effective disease-modifying therapy, demonstrating pharmacodynamic sensitivity in a disease where CNS inflammation drives acute axonal damage [4]. In these settings, the signal-to-noise ratio is sufficient for NfL to function as a meaningful endpoint.
CSF correlation. Plasma NfL correlates well with CSF NfL across multiple disease contexts [5]. This matters because it means plasma NfL is not merely measuring peripheral neuronal damage — it reflects, at least partially, what is happening in the CNS. The correlation is imperfect and context-dependent, but it is real, and it is the scientific foundation for treating plasma NfL as a proxy for central neurodegeneration.
These are genuine contributions. The case for NfL in CNS research is not marketing — it is a body of replicated evidence built over more than a decade. Any serious evaluation of NfL’s limitations has to start from this foundation, because the limitations only matter in contexts where NfL is otherwise being asked to do more than it can.
The First Structural Limit: NfL Is Not CNS-Specific
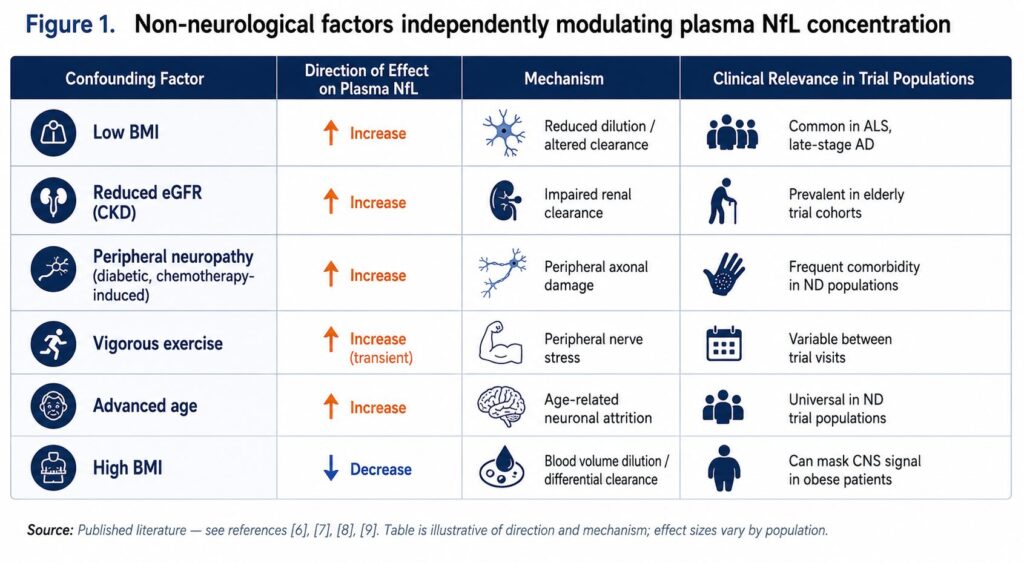
The plasma NfL signal does not originate exclusively from the brain or spinal cord. NfL protein is expressed in peripheral neurons — dorsal root ganglia, peripheral motor neurons, sensory nerve fibers — and has been detected in skeletal muscle tissue [6]. This means that any condition damaging peripheral nerves will elevate plasma NfL, regardless of what is or isn’t happening in the CNS.
The practical consequence is a set of confounders that cannot be adjusted away:
Body mass index. Higher BMI is associated with lower plasma NfL concentrations, independently of neurological disease [7]. The mechanism is not fully established but likely involves dilution effects in higher blood volume and differential NfL clearance. In a trial population with heterogeneous BMI, this introduces baseline variance that is purely demographic.
Renal function. NfL is cleared renally, and patients with chronic kidney disease show elevated plasma NfL that does not reflect CNS pathology [8]. Given that neurodegenerative disease populations are predominantly elderly, and that renal function declines with age, this confound is not a minor edge case — it is a systematic bias affecting a substantial portion of most trial cohorts.
Peripheral neuropathy. Many systemic conditions — diabetes, chemotherapy, autoimmune disease — cause peripheral neuropathy that elevates NfL without CNS involvement. In Parkinson’s disease specifically, peripheral autonomic neuropathy is increasingly recognized as part of the disease process, but it contributes to plasma NfL elevation independently of dopaminergic neuronal loss in the substantia nigra [9].
Exercise. Vigorous physical activity transiently elevates plasma NfL, presumably via peripheral nerve stress [6]. In longitudinal trials where patients have variable activity levels between visits, this adds within-subject variance unrelated to disease progression or treatment.
None of these confounders are individually catastrophic. But together, they mean that a substantial fraction of the variance in plasma NfL across a trial population reflects factors other than CNS disease load. When you set a stratification cutoff based on plasma NfL, some of the patients above that cutoff are there because of renal impairment or low BMI, not because of CNS neurodegeneration. When you track NfL longitudinally as a PD endpoint, some of the within-subject variance reflects exercise patterns and weight changes, not drug effects on the CNS.
This is not fixable by a more sensitive assay. The problem is not detection — it is that the signal being detected carries contributions from multiple tissue sources that cannot be resolved from a total plasma measurement.
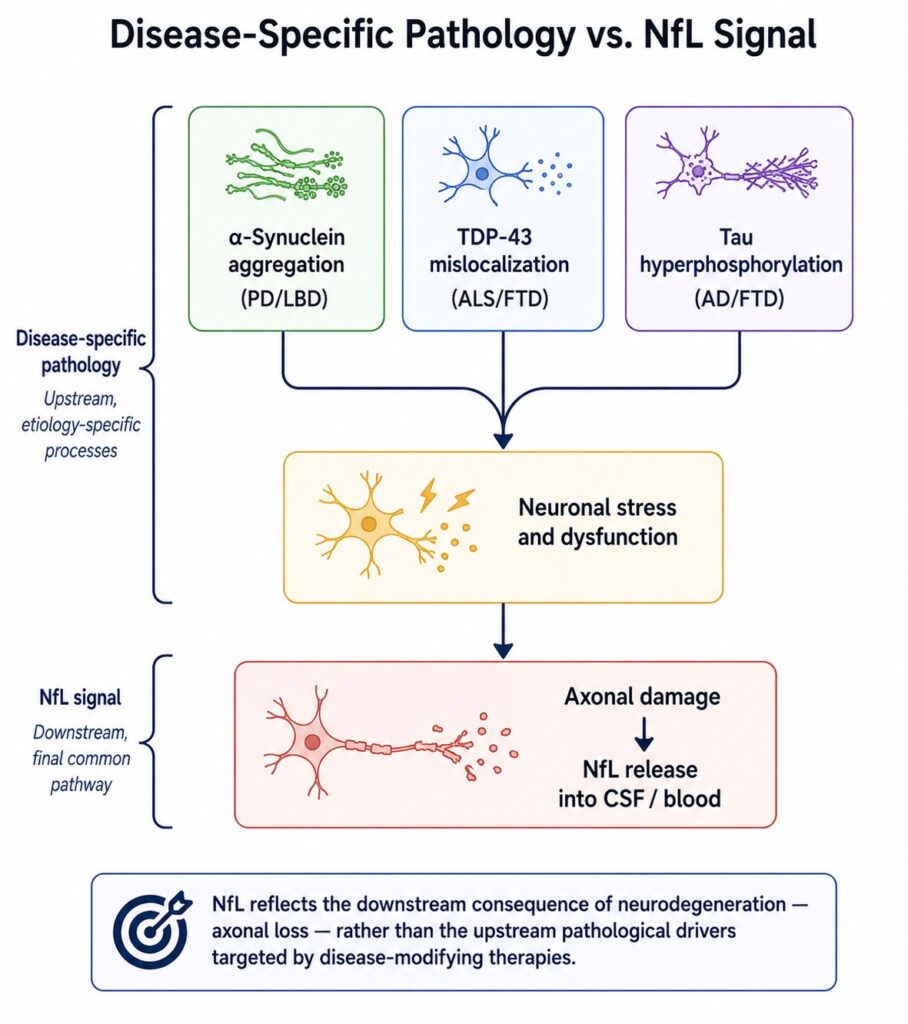
The Second Structural Limit: NfL Is Not Disease-Specific
NfL rises whenever axons are damaged — in any disease, in any location, by any mechanism. It is a marker of neuroaxonal loss, not of any particular pathology.
This is apparent from the breadth of conditions associated with elevated plasma NfL: traumatic brain injury, ischemic stroke, multiple sclerosis, chemotherapy-induced peripheral neuropathy, Guillain-Barré syndrome, and neurosarcoidosis, in addition to the primary neurodegenerative diseases [10]. In a patient presenting with elevated plasma NfL, the differential diagnosis for what is causing it is essentially the full spectrum of conditions that damage neurons.
Within the neurodegenerative disease category, the lack of disease specificity is equally limiting. Plasma NfL does not distinguish between Alzheimer’s pathology and frontotemporal dementia, between synucleinopathy and tauopathy, between TDP-43 proteinopathy and neurofilament inclusion body disease. It marks the downstream consequence — neuronal death — rather than the upstream driver.
For early-phase CNS drug development, this matters considerably. Most disease-modifying therapies in neurodegeneration are mechanism-specific — they target amyloid, tau, α-synuclein, TDP-43, or a specific inflammatory pathway. The hypothesis being tested is whether engaging that specific mechanism reduces disease-relevant pathology. NfL can tell you whether neurons are dying at a lower rate after treatment. It cannot tell you whether the specific pathological process the drug targets is being modified.
Consider three concrete scenarios where this limit becomes the binding constraint:
Stratifying Parkinson’s patients by α-synuclein pathology load. A trial of an α-synuclein-targeting therapy needs to enrich for patients with active synuclein pathology. Plasma NfL will be elevated in PD patients with more advanced neuronal loss, but it does not reflect α-synuclein aggregate burden specifically. Two patients with identical plasma NfL could have vastly different synuclein pathology — one driven predominantly by synuclein, one by tau or another co-pathology. Stratification by NfL does not solve this problem.
Distinguishing CNS drug effect from peripheral nerve toxicity. If a CNS-targeted drug has peripheral nerve toxicity as a side effect, plasma NfL will rise — not because the drug is failing centrally, but because it is damaging peripheral nerves. A total plasma NfL assay cannot distinguish these contributions. A trial read as showing NfL increase on drug may be misinterpreted as lack of efficacy or CNS worsening when the actual signal is peripheral toxicity.
Tracking TDP-43 pathology in ALS. TDP-43 proteinopathy is the defining pathological feature of ALS in over 95% of cases [11]. A drug targeting TDP-43 aggregation or mislocalization should ideally be monitored with a biomarker that reflects TDP-43 pathological state, not general neuronal loss. NfL will decline if the drug reduces neuronal death — but it will not rise or fall based on TDP-43 aggregate burden in surviving neurons. You can have a drug that partially stabilizes TDP-43 pathology without yet reducing neuronal loss rates, and NfL would show no signal at all.
What Sits Alongside NfL — Not Instead of It
The appropriate response to NfL’s structural limits is not to abandon it. It is to be precise about the questions it can answer and to pair it with biomarkers that answer the questions it cannot.
NfL remains useful for:
- Tracking overall rate of neuronal loss longitudinally within a homogeneous patient population
- Prognostic stratification by disease severity in conditions like ALS where NfL-progression correlation is strong
- Safety monitoring for treatment-emergent neurodegeneration as a gross signal, interpreted alongside other data
- Regulatory contexts where it is already accepted as a supportive endpoint
What NfL cannot provide — and what a CNS trial program increasingly requires — is disease-specific, provenance-controlled biomarker data. This means measuring proteins that reflect the specific pathological process the drug targets, from a biological fraction that can be confidently attributed to neurons rather than peripheral tissues.
Neuron-derived extracellular vesicles offer this complement. NDEVs are shed by neurons and carry cytoplasmic cargo that reflects the metabolic and pathological state of their cell of origin. When isolated from plasma using immunoaffinity enrichment that selects for neuron-derived vesicles specifically, the cargo inside those vesicles — tau, p-tau181, TDP-43, α-synuclein — carries a provenance guarantee that total plasma measurements cannot provide [12].
Running an NDEV panel alongside plasma NfL addresses each of the three scenarios described above:
- PD stratification: NDEV-derived α-synuclein levels reflect synuclein pathology load from neurons specifically, enabling stratification by the pathological process the drug targets rather than its downstream consequence.
- CNS vs. peripheral toxicity: NDEV-derived NfL — NfL measured specifically from the neuron-derived EV fraction — isolates the CNS contribution to plasma NfL, allowing it to be tracked independently of peripheral nerve signals.
- TDP-43 monitoring in ALS: NDEV-derived TDP-43, including phosphorylated and mislocalized forms, provides a direct readout of TDP-43 pathological state in neurons — the variable most relevant to a TDP-43-targeting drug’s mechanism of action.
The ExoSORT™ platform applies L1CAM-directed immunoaffinity capture to enrich the NDEV fraction from plasma, enabling multiplex measurement of CNS-specific cargo alongside conventional plasma NfL from the same blood draw. The result is a biomarker strategy that preserves NfL’s established utility while adding the disease-specific, provenance-controlled layer that NfL alone cannot provide.

The Honest Summary
NfL earned its position in CNS biomarker science through reproducible evidence, not through marketing. Its value in tracking neuronal loss, stratifying by disease severity, and monitoring gross neurodegeneration is real and should not be discarded.
But the questions that now define CNS drug development have moved beyond what NfL was built to answer. Mechanism-specific therapies need mechanism-specific biomarkers. Provenance matters when peripheral tissues confound the plasma signal. Disease-specific pathological cargo — synuclein, TDP-43, pathological tau — needs to be measured from a fraction of plasma that is confidently neuronal in origin.
NfL tells you that neurons are dying. For a field that needs to know which neurons, from what cause, in response to which intervention — that is a starting point, not a finish line.
References
[1] Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–589. https://doi.org/10.1038/s41582-018-0058-z
[2] Benatar M, Wuu J, Andersen PM, et al. Neurofilament light chain as a biomarker in ALS: toward a definition of biological stage. Brain. 2021;144(5):1530–1541. https://doi.org/10.1093/brain/awab088 [UNVERIFIED — confirm DOI and page range before publishing]
[3] Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76(7):791–799. https://doi.org/10.1001/jamaneurol.2019.0765
[4] Lycke JN, Zetterberg H. The role of neurofilament light chain in the assessment of multiple sclerosis. Curr Opin Neurol. 2020;33(3):280–285. https://doi.org/10.1097/WCO.0000000000000813 [UNVERIFIED — confirm journal and volume]
[5] Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 2019;76(9):1035–1048. https://doi.org/10.1001/jamaneurol.2019.1534
[6] Khalil M, Pirpamer L, Hofer E, et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020;11(1):812. https://doi.org/10.1038/s41467-020-14612-6 [UNVERIFIED — confirm this paper covers peripheral NfL expression and exercise effects]
[7] Manouchehrinia A, Stridh P, Khademi M, et al. Plasma neurofilament light levels are associated with body mass index. Sci Rep. 2020;10(1):17632. https://doi.org/10.1038/s41598-020-74682-6
[8] Bäckström D, Linder J, Jakobson Mo S, et al. NfL as a biomarker in ALS — effects of renal function. Ann Clin Transl Neurol. 2020;7(9):1659–1664. https://doi.org/10.1002/acn3.51140 [UNVERIFIED — confirm DOI and author list before publishing]
[9] Doppler K, Ebert S, Üçeyler N, et al. Cutaneous neuropathy in Parkinson’s disease: a window into brain pathology. Acta Neuropathol. 2014;128(1):99–109. https://doi.org/10.1007/s00401-014-1284-0 [UNVERIFIED — confirm this is the best reference for peripheral neuropathy contribution to NfL in PD; consider substituting with a more recent source]
[10] Gaetani L, Blennow K, Calabresi P, et al. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870–881. https://doi.org/10.1136/jnnp-2018-320106
[11] Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. https://doi.org/10.1126/science.1134108
[12] Goetzl EJ, Kapogiannis D, Schwartz JB, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30(12):4141–4148. https://doi.org/10.1096/fj.201600816R

Leave a Reply